CHAPITRE 2 : ELABORATION DU REVETEMENT
Les études antérieures montrent qu’un mélange de poudres de HfB2 et de SiC est très difficile à fritter(8, 9, 10, 11). Les procédés utilisés pour élaborer de tels matériaux composites denses font appel au frittage sous charge, à la compression isostatique à chaud ou nécessitent, pour les dépôts chimiques, un contrôle complexe des flux de gaz autour de la pièce à revêtir (31, 32, 17).
Notre objectif est de développer un procédé simple, fiable, utilisable sur des pièces de grandes dimensions et de formes complexes et facile à mettre en oeuvre. Nous discuterons des avantages et inconvénients des techniques employées jusqu’à ce jour, afin d’argumenter le choix du procédé d’élaboration utilisé.
1. LES PROCEDES D’ELABORATION
1.1 Le dépôt chimique en phase vapeur
La codéposition d’un système binaire tel que HfB2-SiC est très difficile à réaliser. Malgré l’étude thermodynamique des conditions de dépôt menée par Lackey et al(32) qui montre la possibilité théorique de réaliser de tels revêtements, les essais d’élaboration restent infructueux. Par exemple, Fenter et al(17), pour un système similaire, obtiennent un mélange ZrB2-B4C au lieu de ZrB2-SiC. Une des solutions envisagées est la superposition de couches de quelques dizaines de microns, des constituants du système. Ainsi, Brockmeyer et al(4) ont élaboré un empilement de couches de HfC et de SiC. Aucune réalisation de composites HfB2-SiC n’a été publiée jusqu’à présent.
1.2 La projection thermique
Les techniques de projection thermique sont particulièrement bien adaptées à l’élaboration de revêtements de matériaux réfractaires. Cependant, les matériaux non-oxydes posent le problème de l’oxydation des particules en vol même en utilisant l’argon comme gaz porteur, par exemple dans une torche à plasma soufflé(2). Il est nécessaire d’effectuer la projection dans une enceinte sous atmosphère contrôlée. L’utilisation d’un simple mélange de poudres de HfB2 et de SiC entraîne un défaut de SiC dans le revêtement final(8, 9, 10, 11). Il est indispensable de préparer des poudres pré-alliées pour projeter simultanément HfB2 et SiC et contrôler ainsi la composition chimique du revêtement final(2).
Ces techniques sont coûteuses et mal adaptées pour des pièces de formes complexes et surtout pour les matériaux sensibles à l’oxydation comme les borures. Une installation sous atmosphère contrôlée est nécessaire, entraînant des limitations de la taille des pièces.
1.3 Le frittage naturel
Le frittage naturel sous atmosphère inerte est le procédé le plus simple. L’utilisation du vide permet de limiter l’effet de contre pression du gaz emprisonné dans la porosité et d’atteindre une densité plus élevée. Cependant, le départ d’espèces volatiles (Si, SiO...), à haute température (8), entraîne une perte de masse importante et une variation de la composition globale. L’argon semble une meilleure solution, malheureusement, les densités obtenues sont inférieures à 90% et peuvent s’expliquer par la présence d’oxygène dans la poudre de départ ou dans l’argon utilisé. De plus, des problèmes d’anisotropie de forme des grains et la présence de zones de forte porosité, qui peuvent provenir de l’évaporation des constituants, limitent l’utilisation de cette technique.
Dans le système ZrB2-SiC, même en utilisant Zr ou ZrH4 comme ajout de frittage, il est très difficile d’obtenir une densité supérieure à 90% sans atteindre 2273K, ce qui implique des températures plus élevées pour le système HfB2-SiC(8, 9, 10, 11).
1.4 Le frittage sous charge
L’application d’une charge lors du frittage permet de combler les pores de plus faible diamètre et d’obtenir une densité proche de la densité théorique. Cette technique se limite à des formes simples et de taille assez réduite car la pression doit être répartie de manière la plus homogène possible dans le matériau.
Malgré les températures et les pressions utilisées, 2473K sous 20MPa, pendant 12000 s(36, 37), des aides au frittage sont nécessaires. Les ajouts utilisés sont Fe, Ni, Co, W, C, WC (17, 29, 8, 9, 10, 11) et les densités relatives obtenues d’environ 98%. Cependant, la taille des grains augmente dans une proportion assez importante.
Cette technique est adaptée à l’élaboration de monolithes mais l’usinage des pièces est très coûteux et la fabrication de revêtements ne peut pas être envisagée.
1.5 Le frittage – réaction
Une alternative intéressante au frittage naturel ou sous charge est le frittage réaction. Il s’agit dans ce cas de former des produits à partir de précurseurs. Deux approches ont été étudiées dans le cas des systèmes Zr(Hf)B2-SiC.
La première consiste à réaliser une réaction en phase solide à partir de zirconium, de silicium et de B4C pour former ZrB2-SiC(48). Le mélange doit être fritté sous argon à 2173K sous une charge de 30MPa pendant 3600s pour obtenir le composite dense.
La seconde approche consiste à synthétiser une phase par réaction chimique autour des grains de la matrice. C’est l’approche choisie par EADS-LV(12). Un mélange de HfB2 et de carbone est mis en présence de silicium gazeux. Du carbure de silicium se forme par réaction avec le carbone. Ce procédé est difficile à maîtriser, il présente de plus les mêmes inconvénients que l’infiltration chimique en phase vapeur (ICPV) lors de l’application à de grandes pièces de forme complexe : la gestion de flux gazeux dans des fours spécifiques.
Une troisième approche concerne l’infiltration d’une préforme de HfB2 par un polysilazane(3) ou du polyméthylsilane(38). Après dégradation thermique à basse température, il se forme des nanoparticules de carbonitrure de silicium avec un dégagement d’hydrogène. L’utilisation d’un précurseur de synthèse permet d’assurer la pureté et la stœchiométrie du carbure de silicium mais des cycles multiples d’infiltration sont nécessaires pour obtenir une bonne densification.
1.6 Conclusions
Pour élaborer un revêtement dans le système Hf-B-Si-C, sur des pièces de grande taille et de forme complexe, le frittage réaction semble offrir des possibilités intéressantes sur le plan industriel. L’agent d’infiltration peut être apporté par voie gazeuse ou liquide. Les travaux déjà menés à EADS-LV sur la voie gazeuse conduisent au développement d’équipements haute température spécifiques qui présentent une complexité équivalente à celle des moyens ICPV. L’infiltration réactive en phase liquide semble apporter une alternative intéressante car le silicium peut être réparti uniformément à la surface de la pièce avant son infiltration.
2. ELABORATION DE LA PROTECTION
Nous avons choisi de développer un procédé céramique par voie liquide pour l’élaboration de revêtements de 200.10-6 à 300.10-6 m d’épaisseur, à partir de poudres mises en suspension. L’élaboration s’effectue en deux grandes étapes. La première est la réalisation d’une préforme poreuse de HfB2 et de carbone. Ce dernier est apporté par un précurseur liquide, une résine phénolique. La seconde étape consiste à effectuer une infiltration réactive de silicium liquide pour former du carbure de silicium autour des grains de HfB2 et à l’interface avec le substrat.
En raison des difficultés pour caractériser les revêtements, des monolithes ont été élaborés en parallèle des revêtements. Ils se présentent sous forme de plaques d‘environ 200.10-6 à 300.10-6 m d’épaisseur. Les monolithes permettront de caractériser la microstructure issue du procédé mis au point et d’étudier le comportement à l’oxydation du matériau.
3. LES MATIERES PREMIERES
3.1 La poudre de HfB2
L'analyse chimique de la poudre commercialisée par Alfa montre la présence de borure de zirconium, comme impureté principale, et de palladium. (CNRS Service Central d’Analyse Vernaison) (tableau 9)
| Elément | Hf | B | Zr | Pd (ppm) |
| Concentration massique (%) | 89 ± 2 | 10,4 ± 0,3 | 1,1 ± 0,3 | 230 |
La distribution de la taille des grains, mesurée par granulomètrie laser en voie liquide (CILAS 1064) fait apparaître une forte agglomération. Le diamètre moyen des agglomérats est de 6-7.10-6 m.
Figure 13 : Distribution granulométrique de la poudre de HfB2
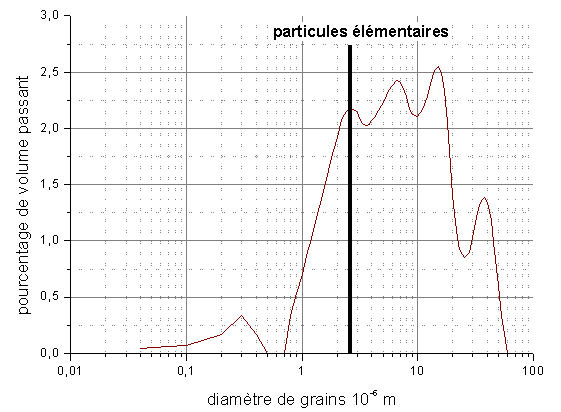
La surface spécifique de la poudre déterminée par la méthode BET (Micromeritics ASAP2000) est de 490 m²/kg. Le diamètre des grains élémentaires supposés sphériques, évalué à partir de cette valeur, est de 1,16.10-6 m, ce qui est en accord avec les observations au microscope électronique (diamètre ∼2.10-6 à 3.10-6 m).
3.2 La poudre de silicium
L'analyse chimique de la poudre de silicium (Alfa) révèle la présence de bore et de zirconium comme impuretés principales.
| Elément | Si | B | Zr | Nd | Pb |
| Concentration massique (%) | 92 ± 3 | 0,14 ± 0,02 | 625(ppm) | 165(ppm) | 250(ppm) |
Le complément à 100% est représenté par l'oxygène issu de l'oxydation du silicium pendant son stockage en présence d'air, soit environ 8%masse.
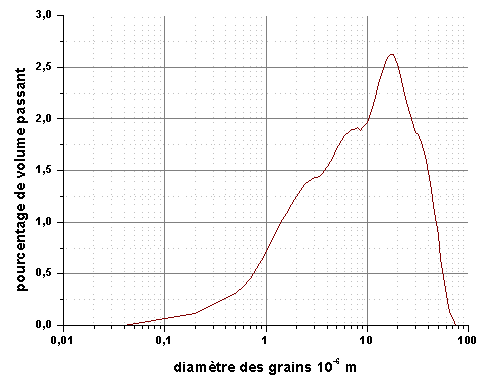
La mesure de la taille des particules montre une distribution très large. La présence d’agglomérats est confirmée par microscopie électronique.
Figure 14 : Distribution granulomètrique de la poudre de silicium
3.3 La résine phénolique
La résine phénolique est une résine de type résol, de faible masse moléculaire, de pH 7,8 et dont la teneur en phénol est comprise entre 19 et 22%. Elle se présente sous forme liquide, de viscosité 0,70-0,85 Pa.s à 293K avec une teneur en eau de 11 à 13%. Le durcissement débute à 353K et le temps de gel à 398K est de 1320 à 1800 s. Cette résine a été choisie pour son rendement élevé en carbone (donnée fournisseur 56%).
3.4 Le liant organique
La carboxyméthylcellulose (CMC) (Aldrich) a été choisie pour la préparation de la suspension de silicium. C’est une solution à 4% d’eau, de viscosité comprise entre 0,01 à 0,055 Pa.s.
3.5 Le dispersant
La mise en suspension de la poudre de HfB2 nécessite le développement d’interactions de surface afin de limiter l’agglomération des grains et de stabiliser cet état.
Le dispersant a été choisi pour son effet stérique car l’effet électrostatique ne se développe pas dans les milieux apolaires comme l’éthanol. C’est un mélange d’esters phosphoriques de masse moléculaire moyenne égale à 700.
3.6 Les substrats
L’essentiel de l’étude a été réalisée sur du graphite (ATJ) comme substrat d’essai pour l’élaboration de revêtements à cause de son faible coefficient de dilatation thermique, proche de celui des composites. Les substrats sont préparés sous forme de cylindres de diamètre 10-2 m et de longueur 2,5.10-2 m. Les extrémités sont usinées pour leur donner une forme sphérique. Ces précautions permettent de minimiser les effets de bords (sources de défauts d’épaisseur dans les procédés d’enduction).
L’applicabilité du procédé a été testée sur des composites utilisés dans le domaine spatial, carbone-carbone et carbone-carbure de silicium avec plusieurs architectures fibreuses : carbone/carbone 2,5D et 3D, à fibres méchées ou continues, et carbone/SiC 2,5D.
4. ELABORATION DE LA PROTECTION
4.1 Essais exploratoires
Des travaux préliminaires avaient été entrepris par EADS-LV(12) sur l’élaboration d’un revêtement HfB2+SiC par siliciuration d’une préforme de HfB2+C. Tout d’abord en phase gazeuse, soit à partir des vapeurs d’un bain de silicium liquide, soit par cémentation, à partir d’un mélange SiC-SiO2.
En ce qui concerne la voie liquide, trois poudres de HfB2, de granulométries comparables, deux poudres de silicium et deux sources de carbone ont été testées. Il semble que la nature des poudres de HfB2 ait peu d’influence sur la microstructure du revêtement. Cependant, la forme sous laquelle est introduit le carbone, soit en poudre, soit par dégradation d’une résine phénolique, conduit à des microstructures différentes. L’utilisation de résine phénolique diminue la taille des particules de carbone et améliore l’homogénéité du mélange HfB2-SiC.
En effet, les microstructures après siliciuration montrent une répartition assez hétérogène de HfB2 dans le revêtement avec des agglomérats de plusieurs dizaines de microns. Deux types de microstructures sont obtenus (figure 15). La première se caractérise par un revêtement dense, riche en silicium libre, très adhérent et présente un grossissement des grains de HfB2 (figure 15a). La seconde est poreuse, exempte de silicium libre, faiblement adhérente et sans grossissement des grains de HfB2 (figure 15b). Aucune influence de la quantité de silicium ou de la température de traitement sur la microstructure n’a été établi. Il est à noter que la structure poreuse présentée sur la figure 15b) a probablement subi un arrachement des grains lors de la préparation de l’échantillon. La porosité du revêtement est, en fait, plus faible que ne le laisse supposer la micrographie.
Le niveau de connaissance du procédé à ce stade exploratoire ne permettant pas de maîtriser l’orientation sur un type de microstructure, ce point constituera un des objectifs de ce travail.
Figure 15 : Micrographies des sections polies de revêtements siliciurés sous argon. a) structure dense b) structure poreuse

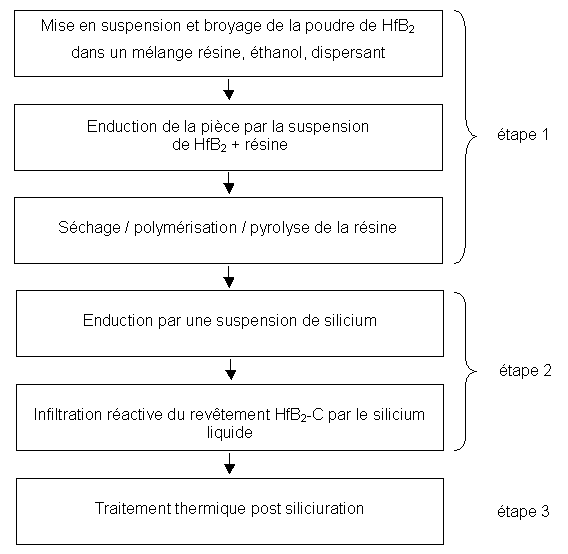
4.2 Synoptique du procédé d’élaboration
Le procédé d’élaboration se décompose en trois étapes :
- la réalisation d’une préforme poreuse constituée de HfB2 et de carbone à partir d’une suspension de poudre de HfB2 et de résine phénolique.
- l’infiltration réactive de silicium liquide pour former du carbure de silicium autour des grains de HfB2 à l’interface avec le substrat.
- un traitement thermique optionnel en fonction de l’application.
Ces trois étapes de décomposent en 6 phases présentées sur le synoptique ci dessous (figure 16).
Figure 16 : Synoptique du procédé d'élaboration
4.3 Elaboration de la structure poreuse HfB2-carbone
L’objectif de cette étape est d’élaborer une couche constituée de HfB2 et de carbone, qui sera traitée thermiquement en présence de silicium liquide pour former le mélange HfB2-SiC. Il faut donc contrôler la distribution de la taille des pores, la composition chimique et l’homogénéité de répartition des phases.
Pour cela, il est nécessaire d’optimiser la granulométrie de la poudre de HfB2 pour obtenir un revêtement assez dense et une répartition homogène. Il est également important de stabiliser l’état de dispersion de la suspension, car HfB2 est très dense et sédimente très vite. De plus, il faut ajuster la viscosité de la barbotine afin d’obtenir un revêtement d’épaisseur voulue, d’incorporer la quantité de résine phénolique nécessaire à l’obtention de la composition chimique souhaitée, de s’assurer que la résine ne forme pas une émulsion dans la barbotine et de déterminer les paramètres qui imposent la porosité du mélange HfB2+carbone.
4.3.1 La suspension de HfB2-résine - Enduction des substrats
4.3.1.1 Le broyage de la poudre de HfB2
L’étude préliminaire a mis en évidence l’hétérogénéité de la répartition des grains de HfB2, qui peut être attribuée à la forte agglomération de la poudre de départ, avec une taille maximale des agglomérats d’environ 60.10-6 m. De plus, un bon empilement des particules est favorable à l’obtention d’un revêtement compact.
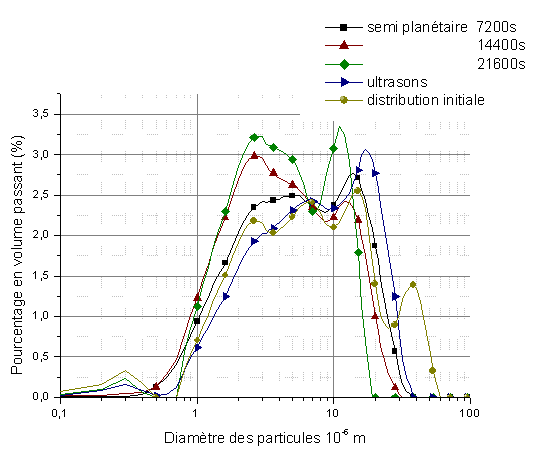
Nous avons cherché à détruire ces agglomérats par les ultrasons. Ce traitement ne s’avère pas assez efficace car il ne permet que de casser les agglomérats durs d’une taille supérieure à 30.10-6 m alors que les particules élémentaires ont un diamètre de 10-6 à 3.10-6 m (figure 17). L’utilisation d’un broyeur semi-planétaire avec des billes de carbure de tungstène, plus énergétique, est donc nécessaire.
Il est également indispensable de stabiliser l’état de désagglomération des particules de HfB2 pendant la durée d’élaboration du revêtement. Des travaux sur la préparation de suspensions de composés du bore, comme ZrB2 (masse volumique 6090 kg/m3), ont montré la difficulté d’étudier de telles suspensions à cause de la densité élevée des poudres(27). Celle de HfB2 (10500 kg/m3) ne permet pas l’utilisation de certaines techniques classiques de caractérisation des dispersions telles que l’acoustophoromètrie, la viscosimètrie... à cause d’une sédimentation trop rapide. Cependant, cette propriété peut être mise à profit pour évaluer la qualité de la dispersion. Le rapport entre la hauteur du liquide surnageant et celle du sédiment donne une indication sur la qualité de l’empilement des particules, qui est liée à la distribution granulométrique de la poudre.
L’étude de la suspension a été réalisée en mesurant l’évolution de la distribution de la taille des particules de HfB2, par granulométrie au laser, et la hauteur de sédimentation de la suspension en fonction du temps de broyage. La quantité de dispersant nécessaire à la stabilisation est déduite de la surface spécifique de la poudre de HfB2, et de l’expérience du laboratoire acquise sur l’utilisation de ce dispersant.
La poudre de HfB2, l’éthanol et le dispersant sont introduits et broyés pendant 7200, 14400 ou 21600 s dans une jarre en polyéthylène contenant des billes de carbure de tungstène. La résine est ajoutée à la fin du broyage pour éviter tout endommagement des chaîne polymériques.
Le suivi de l’évolution de la distribution de la taille des grains de HfB2 montre que le broyage semi-planétaire est plus efficace que la dispersion aux ultrasons (figure 17). La taille maximale des agglomérats est de 20.10-6 m avec une augmentation de la proportion des particules élémentaires, 10-6 à 3.10-6 m. Le diamètre à 50% des grains passe de 7.10-6 m à moins de 4.10-6 m, avec une distribution légèrement moins dispersée.
Figure 17 : Influence du broyage sur la distribution granulométrique de la poudre de HfB2
Le tableau 11 montre l’évolution du rapport hauteur de liquide surnageant sur hauteur de sédiment après 3 jours de sédimentation. Le bénéfice qu’apporte la technique et le temps de broyage est très marqué. Il semble que la quantité de dispersant ne dépasse pas la valeur critique, qui conduit à la réagglomération des particules, à cause de la diminution locale de la quantité de phase liquide.
Tableau 3 : Evaluation de la qualité de la dispersion par sédimentation après 3 jours de sédimentation

Un autre point important mis en évidence par l’étude préliminaire est la reproductibilité de la microstructure. Il est nécessaire de s’assurer qu’il est possible de reproduire chaque étape pour espérer contrôler la microstructure finale.
Les mesures de la distribution de la taille des grains ont été réalisées systématiquement sur cinq lots de barbotine. La figure 18 montre une bonne reproductibilité de l’étape de broyage avec de légères fluctuations de la proportion des agglomérats les plus gros, pour les temps de broyage les plus longs.
Figure 18 : Etude de reproductibilité de la distribution granulométrique de la poudre de HfB2 après un broyage semi-planétaire de 21600s.

4.3.1.2 Protocole expérimental
La barbotine est réalisée par broyage dans une jarre en polyéthylène du mélange de poudre de HfB2, d'éthanol et de dispersant. A l’issue de 21600 s de broyage, la résine phénolique est introduite dans la jarre et le mélange est homogénéisé.
Deux types d’échantillons sont élaborés à partir de la barbotine. D’une part les revêtements sur substrats carbonés et d’autre part les monolithes.
Les substrats en graphite, composite carbone/carbone et carbone/carbure de silicium sont nettoyés et séchés pour éliminer les particules de carbone présentes en surface.
L’enduction est réalisée par immersion des substrats dans la suspension et les monolithes par coulage de la barbotine sur un papier anti-adhésif.
4.3.2 Polymérisation et pyrolyse de la résine précurseur de carbone
La résine phénolique doit être traitée thermiquement pour former du carbone. Cette décomposition s’effectue en deux étapes, la première est la polymérisation et la seconde la pyrolyse. Ces phénomènes s’accompagnent de dégagements gazeux qui sont à l’origine du développement de la porosité.
Après avoir rappelé les processus chimiques mis en jeu dans la thermolyse de la résine, nous étudierons l’influence du cycle thermique sur la microstructure du mélange HfB2+carbone.
4.3.2.1 La dégradation thermique des résines phénoliques
Les résines phénoliques sont le résultat de la polycondensation du formaldéhyde et du phénol. Une grande variété de structures et de propriétés sont obtenues en fonction des produits de départ et des traitements thermiques. Les résines phénoliques ont été choisies pour leur bon rendement en carbone après pyrolyse sous atmosphère inerte.
Nous utilisons une résine de type résol, thermodurcissable, soit un mélange formaldéhyde-phénol dans un rapport molaire de 1,5. Dans ces conditions, la concentration en sites réactifs est suffisante pour que la polycondensation se produise par simple chauffage. Le matériau obtenu est insoluble et infusible. Sa structure dépend de la nature de la catalyse (acide ou basique), du rapport entre les réactifs et de la température de traitement. Le mécanisme de la polycondensation est présenté à la figure 19. L’eau libérée pendant la réaction peut conduire à la formation d’une large porosité, si la vitesse de montée en température est trop élevée ou si l’épaisseur de la pièce est trop grande. Les cycles couramment utilisés ont une vitesse de montée en température de quelques degrés par heure pour des épaisseurs de plusieurs millimètres.
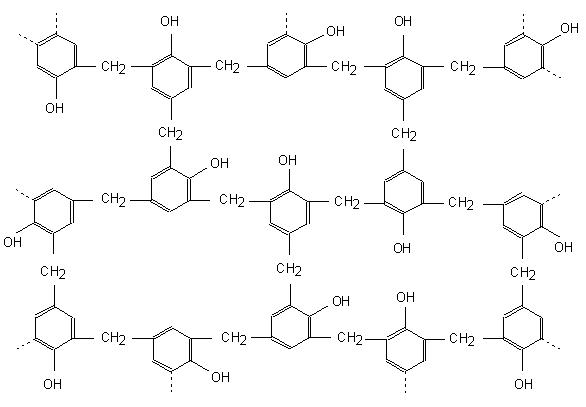
Une structure possible de la résine après polymérisation en milieu basique est présentée à la figure 20.
Figure 19 : La polycondensation d’une résine phénolique de type résol(23)
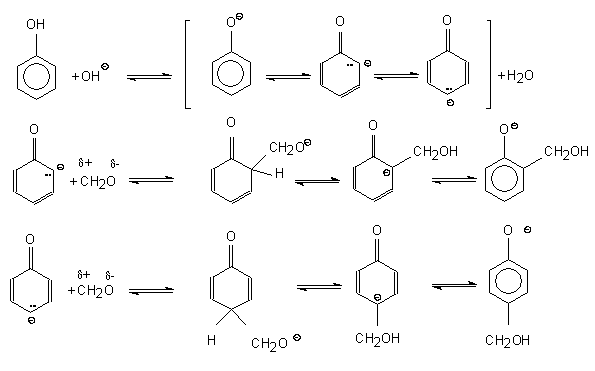
Figure 20 : Structure possible de la résine phénolique polymérisée(19)
La pyrolyse de la résine phénolique polymérisée s’accompagne également de dégagements gazeux qui forment une porosité dont la distribution en taille dépend des conditions expérimentales et de la morphologie de l’échantillon. A partir de 473K, une importante quantité d’eau est éliminée. Il a été montré qu’elle est plus importante sur des échantillons pulvérulents que sur des massifs(33). Puis entre 473 et 673K, ce sont essentiellement CO et CO2 qui sont émis. Au-delà de 673K, les réactions de déshydratation, de déshydrogénation et de crackage de la résine débutent, conduisant à la libération de CH4 et H2. Au-dessus de 1023K, seul l’hydrogène est détecté. Des réactions de condensation se produisent pour donner des polycycles aromatiques.
Les pores formés après une montée lente sont très petits (0,1.10-6 m) et répartis de manière homogène alors qu’une montée rapide entraîne la formation d’une structure hétérogène constituée de pores plus gros. Les auteurs indiquent des vitesses critiques de montée en température pour la transition entre ces phénomènes, de l’ordre de 1,67°/s pour des échantillons massifs de 10-3 m d’épaisseur(7).
Une augmentation de la température de traitement entraîne la coalescence des pores, d’où un élargissement de la distribution en taille de la porosité et un diamètre moyen des pores plus grand.
La structure du carbone obtenu par pyrolyse sous azote à 1273K avec une vitesse de montée en température de 2,77.10-2°/s, est constituée de globules résultant de l’empilement de plans graphitiques(42). La réactivité de ce carbone est grande compte tenu de la surface relative des côtés de plans graphitiques par rapport à celle des faces.
4.3.2.2 Protocole expérimental
La polymérisation est réalisée dans les conditions habituelles utilisées par l’industriel pour de tels produits. Puis le polymère est pyrolysé sous atmosphère inerte.
4.3.2.3 Influence de la vitesse de montée en température
La vitesse de montée en température peut influencer la distribution en taille de la porosité, qui est un facteur important pour la siliciuration ultérieure. L’autre paramètre est l’épaisseur de résine à traiter. Plus elle est fine, plus la vitesse de montée en température pourra être grande.
Deux vitesses moyennes extrêmes, de montée en température, ont été étudiées. La première, 8,33.10-4°/s correspond à des cycles utilisés pour polymériser des échantillons de plusieurs millimètres d’épaisseur, la seconde, 0,05°/s est rapide.
La figure 21 montre la distribution de la taille des pores de monolithes élaborés dans les conditions précisées ci dessus. La vitesse de montée en température ne semble pas avoir d’influence sur la distribution de la taille des pores.
Figure 21 : Distribution de la taille des pores de monolithes polymérisés et pyrolysés avec des vitesses de montée en température de 8,33.10-4°/s et 0,05°/s
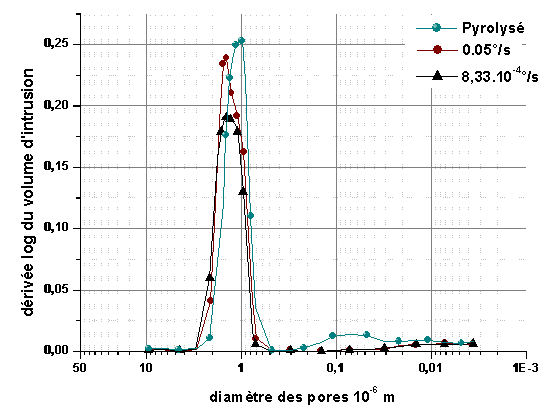
La résine phénolique étant thermodurcissable, on peut supposer que la structure est figée après la polycondensation, jusqu’à la dégradation complète en carbone. L’évolution de la distribution de la taille des pores après pyrolyse n’est pas significative par rapport à l’état polymérisé. L’influence du cycle de pyrolyse sur la structure poreuse semble donc négligeable pour les faibles épaisseurs mises en jeu (figure 21).
4.3.3 Caractérisation des dépôts et des monolithes
Les propriétés importantes pour les traitements ultérieurs sont la structure poreuse et la composition chimique du mélange HfB2+carbone. Le substrat est source d’interférence avec les caractéristiques du revêtement. Pour cette raison, toutes les analyses ont été réalisées sur des monolithes. L’observation de sections polies de revêtements à l’état pyrolysé a permis de rendre compte de l’arrangement des phases (figure 22), mais les difficultés de préparation des échantillons, à cause de leur friabilité, ne permettent pas de réaliser des analyses quantitatives.
La description de la porosité a été obtenue par porosimètrie au mercure (Micromeritics autopore II 9220) et surface spécifique BET (Micromeritics ASAP 2010). Des mesures de densité par picnomètrie à l’hélium (micromeritics Accupyc 1330) ont permis de vérifier ces valeurs. Le rendement en carbone de la résine a été déterminé par analyse chimique (CNRS Vernaison). Plusieurs lots de matériaux ont été étudiés afin de s’assurer de la reproductibilité des résultats et du procédé d’élaboration.
Le rendement en carbone de la résine est assez reproductible avec une valeur moyenne de 35% (tableau 4 et tableau 5). La masse volumique théorique du mélange HfB2-carbone réalisé dans les proportions de l’étude est 8560 kg/m3, en supposant que la masse volumique du carbone vitreux soit égale à 2000 kg/m3. Ceci indique que la porosité est essentiellement ouverte et interconnectée et que la porosité fermée est négligeable (tableau 6 et tableau 7).
| Lot | 2 | 3 | 4 | 5 |
| Rendement en carbone (% en masse) |
38,3 | 32,9 | 35,2 | 34,1 |
| lot | %masseC | %masseHf | %masseB |
| 2 | 5,4 | 81.0 | 9,5 |
| 3 | 4,5 | 81,9 | 9,6 |
| 4 | 4,9 | 79,7 | 9,6 |
| 5 | 4,7 | 82,1 | 9,9 |
| Mode de broyage de la poudre |
Densité squelettique des monolithes | |
| Porosimètrie au mercure | Picnomètrie à l'hélium | |
| Ultrasons | 7,8 | 8,5 |
| Broyage semi-planétaire 14400s | 8,6 | 8,5 |
| Broyage semi-planétaire 21600s | 8,2 | 8,7 |
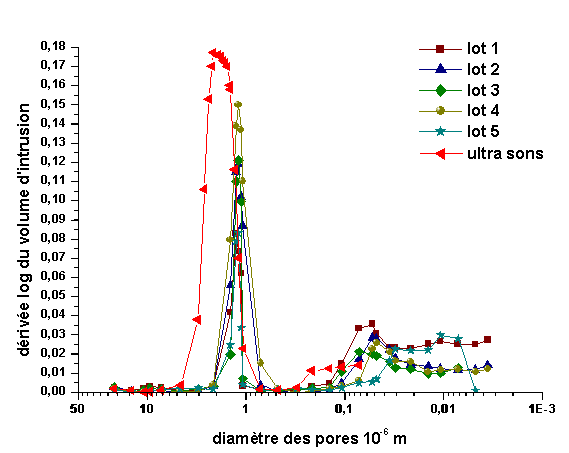
La distribution de la taille des pores pour les différents échantillons est reproductible (figure 23). Les pores peuvent être séparés en deux classes. La première est centrée autour de 10-6 m, avec une distribution assez étroite. La seconde regroupe les pores de taille inférieure à 0,1.10-6 m, avec une large distribution. Le diamètre moyen passe de 1,77.10-6 m après dispersion aux ultrasons à environ 10-6 m après broyage semi-planétaire. La surface spécifique développée après pyrolyse (tableau 8) est largement supérieure à celle de la poudre de HfB2 initiale (490 m²/kg). Ces mesures mettent en évidence la contribution du carbone à la surface spécifique et à la porosité ouverte totale du mélange HfB2+carbone. La porosité la plus large, centrée sur 10-6 m, est liée à l’empilement de particules, comme le montre son évolution en fonction de la technique de broyage utilisée, alors que la microporosité est directement liée aux départs gazeux lors du traitement de la résine. Cette grande surface spécifique augmente la réactivité avec le silicium lors de son infiltration.
| lot | 1 | 2 |
| porosité ouverte totale (%) | 16,6 | 20,5 |
| Lot | 1 | 4 | 5 |
| Surface spécifique (m²/kg) | 22900 | 22600 | 23100 |
Figure 22 : Observation au MEB d’une coupe d’un revêtement HfB2+C pyrolysé

Figure 23 : Influence de la technique de broyage sur la distribution de la taille des pores et reproductibilité de la distribution, après pyrolyse, de monolithes HfB2+C
4.3.4 Discussion et conclusions
La porosité du compact granulaire HfB2+carbone provient de deux sources, l’empilement des particules de HfB2 et les dégagements gazeux qui accompagnent la dégradation de la résine phénolique.
L’optimisation de la distribution de la taille des particules de HfB2, par broyage semi-planétaire, permet de diminuer la distribution en taille de la microporosité du matériau final qui est centrée autour de 10-6 m. Les cycles thermiques conduisant à la polycondensation et à la pyrolyse semblent avoir un effet négligeable sur la porosité totale.
La faible quantité de résine nécessaire à l’obtention de la composition chimique finale de la protection, entraîne que l’épaisseur de résine autour des particules de HfB2 est très faible. La vitesse critique de montée en température lors de la polycondensation, qui sépare les domaines d’obtention d’une large porosité, à cause du départ rapide d’une grande quantité de gaz, d’une porosité fine, est donc très élevée. La résine phénolique étant thermodurcissable, la structure formée par la polycondensation est figée jusqu’à la dégradation thermique en carbone. Ceci implique que le volume poreux n’est pas lié à la présence de carbone mais à la qualité de l’empilement des particules de HfB2. Le rendement en carbone diminue avec l’épaisseur de résine traitée(19), ce qui pourrait expliquer que la teneur après pyrolyse soit plus faible que le taux de coke attendu. Cependant, le rendement est suffisamment reproductible pour pourvoir ajuster la composition du mélange HfB2-SiC.
Au cours de cette expérimentation nous avons recherché un compromis entre amélioration de la compacité des particules de HfB2 recouvertes de carbone et le diamètre des pores afin d’obtenir une distribution homogène de HfB2 et SiC après infiltration et réaction avec le silicium liquide.
4.4 Enduction par une suspension de silicium et infiltration réactive
Les objectifs sont de former une matrice homogène de SiC autour des grains de HfB2 et une couche d’accrochage de SiC à l'interface avec le substrat par réaction entre le silicium liquide et le substrat carboné.
L’infiltration d’un liquide à travers une préforme poreuse a été largement étudié dans le cadre de l’élaboration du carbure de silicium ou de composites SiC-Si(46, 34) et SiC-SiC(38, 28). Les paramètres mis en jeu sont nombreux et complexes et peuvent se classer en quatre catégories, les caractéristiques de la microstructure et la quantification de son évolution au cours du traitement, la réaction chimique entre le liquide et son environnement, les mécanismes d’infiltration, et enfin les propriétés physiques du liquide infiltrant en fonction des conditions de traitement.
Ces grandeurs interagissent largement les unes avec les autres, ce qui rend difficile la modélisation du phénomène dans son ensemble.
L’infiltration réactive sera abordée d’un point de vue théorique. Puis, l’étude des paramètres du traitement de siliciuration permettra de définir un cycle de siliciuration adapté. Enfin, nous présenterons la caractérisation microstructurale et chimique des matériaux obtenus.
4.4.1 L’infiltration réactive
L’infiltration de silicium dans une préforme poreuse de carbone met en jeu trois phénomènes transitoires. Le silicium migre dans la porosité puis réagit avec le carbone avec une augmentation de volume qui entraîne une variation de la distribution de la taille des pores. Cette réaction est exothermique, ce qui modifie les propriétés physiques du silicium et la cinétique de l’infiltration. La mouillabilité du silicium évolue également car la transformation du carbone en carbure de silicium modifie les propriétés de surface et l’augmentation de température diminue l’angle de mouillage.
Après avoir décrit la mouillabilité du silicium, les conditions d’infiltration spontanée et les mécanismes de réaction entre le silicium et le carbone, nous étudierons les modèles d’infiltration réactive développés et leurs limitations.
4.4.1.1 Mouillabilité du Si sur le C et le SiC
La mouillabilité d'un liquide sur une surface dépend de nombreux facteurs, tout d’abord intervient l'homogénéité de la surface. La transposition de la valeur mesurée sur une surface de graphite plane et homogène ne sera pas possible dans le cas d'une préforme poreuse, qui ne pourra pas être considérée comme une surface homogène. Ensuite, la tension de vapeur du liquide en fusion dans l'atmosphère de mesure, doit atteindre un équilibre. Puis, les réactions chimiques modifient la nature de la surface et donc son énergie. Enfin, le temps de mesure doit être constant car l'angle de mouillage évolue avec celui-ci. De plus, le mouillage augmente avec la température.
L’objet de cette étude est la mouillabilité d’un système réactif dans des conditions idéales et l’étude de l’influence de la forme de la porosité sur les forces capillaires.
Un autre paramètre important à prendre en considération est la composition du liquide. Le carbone est soluble en faibles proportions dans le silicium mais contribue à la diminution de l’angle de mouillage.
On considère un système triphasé, liquide, solide et gaz. Les hypothèses sont les suivantes :
- Les forces de gravité sont négligeables,
- La pression et la température sont constantes,
- La courbure n'a pas d'influence sur la pression,
- Les tensions interfaciales sont indépendantes de l'orientation.
- L'énergie libre du système peut alors s'écrire(1) :
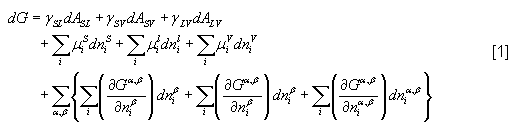
avec i le nombre de composés,
γ la tension de surface entre les phases considérées,
A l’aire de l’interface entre les phases considérées,
G l’enthalpie libre du système,
ni le nombre de moles du composé dans une phase= concentration*volume de la phase,
µi le potentiel chimique du composé i,
α et β les régions homogènes du système.
Le premier terme est relatif à l'équilibre mécanique au niveau des interfaces, le second à l'équilibre chimique des phases. Le troisième terme prend en compte les changements de phase aux interfaces.
Dans le cas d'un mouillage entre deux phases non réactives et à l'équilibre thermodynamique, les termes chimiques sont nuls. La dérivée du terme mécanique permet de déterminer les conditions d'étalement du liquide sur le solide. A partir d'une goutte sphérique, la diminution de l'énergie libre entraîne une augmentation de l'aire de l'interface liquide-solide alors que l'aire de l'interface liquide-vapeur passe par un maximum, diminuant pour θ compris entre 90 et 180° et augmentant pour θ compris entre 0 et 90°.
Dans les conditions d'équilibre une estimation de l'angle de contact peut être obtenue à partir de l'équation :
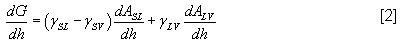
avec h la hauteur de la goutte
A l’aire interfaciale
γ la tension de surface
Une valeur négative de dG/dh indique une diminution continue de la tension de surface et donc le mouillage du solide par le liquide (figure 24).
Dans le cas d’un système réactif, il y a modification des tensions de surface et donc de l’angle de mouillage, avec la réaction. Le transfert de masse qui a lieu a des effets beaucoup plus importants qu’une simple adsorption de molécules car il peut entraîner des variations de volume et de composition.
L’énergie libre de la réaction est constituée de deux composantes. La première est l’énergie libre de fusion, qui ne peut être prise en compte dans le mouillage que lorsque toute la matière est fondue et dépend donc de sa quantité. La seconde est l’énergie libre de réaction. La réaction entre le silicium et le carbone est fortement exothermique. La formation d’une couche de carbure de silicium à la surface du carbone contribue à la diminution de l’angle de mouillage, car la tension de surface du silicium sur le carbure de silicium est inférieure à celle du silicium sur le carbone.
La diminution de l’énergie libre du système est la force motrice du transfert de masse à travers une interface. A l’instant initial, on peut supposer que seule l’interface est concernée. Expérimentalement, des tensions de surface négatives peuvent en résulter, conduisant soit à un mouillage spontané, soit à la formation d’une émulsion. Avec le temps, la réaction se poursuit et les gradients de composition diminuent. Le troisième terme de l’équation [1] décroît et les tensions de surface atteignent une valeur d’équilibre (figure 24).
Dans le cadre d’un système réactif deux équilibres sont envisageables, soit entre la vapeur et le solide soit entre la vapeur et le liquide (figure 25).
Si la vitesse de croissance de la phase interfaciale entre le solide et le liquide est inférieure à celle de l’étalement du liquide, γSV0 ne varie pas, alors que γSL0 est modifiée de la valeur ΔgSL qui est l’enthalpie libre de la réaction entre le solide et le liquide (figure 25). Si γSL0+ ΔgSL est supérieur à γLV, il y a étalement. Dans le cas contraire, l’angle de mouillage diminue jusqu’à atteindre un équilibre. Ensuite, la diffusion dans le solide se poursuit et la force de mouillage est diminuée de ΔgSL d’où une augmentation de θ. Pendant cette augmentation, si la quantité de liquide a fortement diminué, il peut se produire une séparation en plusieurs gouttes.
Si la vitesse de croissance de la phase interfaciale est très grande par rapport à la vitesse d’étalement du liquide, γSV et γSL vont être diminuées toutes les deux de ΔgSL car le liquide est en contact avec le solide qui a réagi. Il n’y aura pas de modifications par rapport au comportement initial.
De nombreux paramètres influencent la mouillabilité des liquides sur les solides. L’étude de ces phénomènes est difficile à transposer à des systèmes plus complexes, c’est à dire multiphasés, non plans et contenant des impuretés.
figure 24 : Variation de la tension de surface entre deux phases qui réagissent (1)
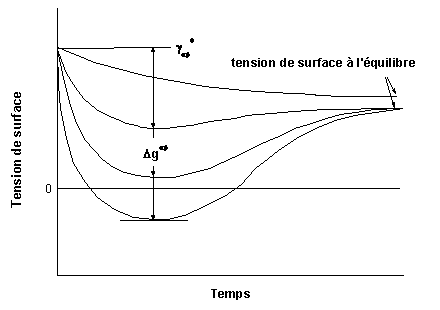
figure 25 : Représentation de l’évolution du mouillage dans le cas d’un système réactif :
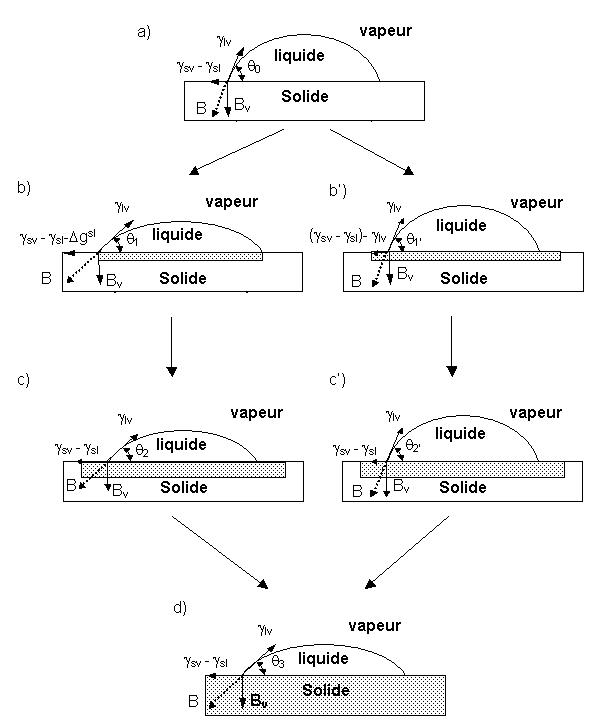
(abcd) si la vitesse de croissance du produit de réaction est inférieure la vitesse d’étalement du liquide
(ab’c’d) dans le cas contraire.
Best la force égale et opposée à γlv et Bv est la composante verticale(1)
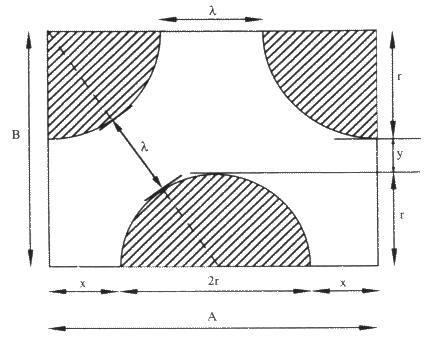
Trumble(47) a étudié les conditions d’infiltration spontanée, par capillarité, de liquides dans une préforme poreuse constituée de particules sphériques. Elles sont discutées en fonction de la taille de la porosité et de la mouillabilité du liquide.
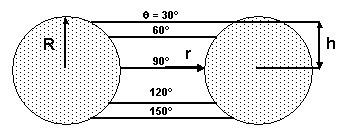
La porosité d’un compact de particules consiste en un réseau de pores ouverts interconnectés. Les liaisons entre les pores ne sont pas cylindriques mais de forme toroïdale (figure 26). Sans pression appliquée (ΔP=0), la position de la surface du liquide à l’équilibre dépend de l’angle de mouillage θ. Un angle de mouillage inférieur à 90° est nécessaire à la pénétration du liquide. La hauteur de pénétration spontanée dépend de l’angle de mouillage (h=R cos θ).
Figure 26 : Section d’un pore de forme toroïdale (R/r=1) avec la pénétration d’un liquide d’angle de mouillage θ (47)
Le calcul plus précis des conditions d’infiltration spontanée dans le cas d’un compact suppose la prise en compte de la taille et de la forme des interstices entre les grains. Pour modéliser plus précisément la porosité d’un compact, il faut considérer une particule située à une distance inférieure à R et des interstices tétraédriques et octaédriques (figure 27).
Figure 27 : Pores tetraédriques (AA) et octaédriques (BB)(47)
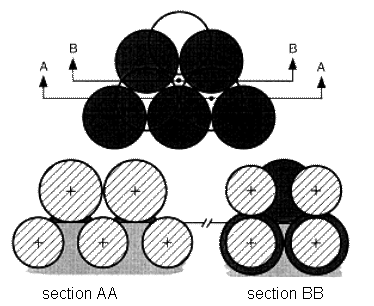
Considérons un empilement de sphères de rayon R, l’infiltration de plusieurs couches nécessite que le liquide pénètre suffisamment pour toucher les sphères de la couche suivante. L’angle critique d’infiltration a été calculé à partir de l’espace entre deux couches de particules et le rayon des sphères, il est θc=50,7°. Il est le même pour les pores octaédriques et tétraédriques (figure 27). En suivant le même raisonnement, on obtient une valeur de θc=65,5° pour infiltrer la première couche. Si les pores sont petits, l’effet de la pesanteur sur la forme du ménisque peut être négligé. Les effets de la distribution en taille des pores et des hétérogénéités nécessitent une étude plus fine pour se rapprocher des conditions réelles.
Les angles de contact du silicium liquide sur le carbone et le carbure de silicium sont des caractéristiques essentielles à la compréhension des phénomènes mis en jeu. Malheureusement, leur détermination est rendue difficile par l’évolution de la nature de la surface liée à la réaction, la variation de la température et la rugosité de la surface. Dans tous les cas, un bon mouillage est nécessaire pour réaliser une infiltration spontanée de la préforme.
4.4.1.2 La réaction entre le silicium et le carbone
La réaction entre le silicium et le carbone a fait l’objet de nombreuses études et plusieurs mécanismes sont avancés :
- La formation quasi instantanée d’une couche de carbure de silicium suivie d’une croissance contrôlée par un régime diffusionnel. (16, 18)
- La formation quasi instantanée d’une couche de carbure de silicium qui s’écaille, en raison de la différence de volume entre le carbone initial et le carbure de silicium, permettant un accès continu au carbone sous-jacent.(24)
- La dissolution du carbone dans le silicium et la précipitation de carbure de silicium quand le liquide est sursaturé.(39)
Fitzer et al(18) ont étudié la réactivité de différentes formes de carbone avec le silicium liquide. Les paramètres majeurs dans les réactions sont la porosité et la microstructure du carbone. Les phénomènes impliqués sont essentiellement liés à la diffusion en volume, par capillarité, du silicium liquide, due à son excellente mouillabilité sur le carbone pur, et à la diffusion aux joints de grains et en volume à travers la couche de carbure de silicium formée.
Des mesures par analyse thermique différentielle réalisées par Singh et al(44) ont montré que la réaction est très rapide et fortement exothermique. La température atteinte semble directement liée aux vitesses d’avancement des fronts de réaction et d’infiltration, donc à la taille de la porosité. Un écart de 873K a été observé entre la température au cœur du matériau et la température de consigne(39). Un large diamètre des pores (10-6 à 2.10-6 m) favorise l’infiltration par rapport à la réaction qui ne peut plus être contrôlée. Cette configuration conduit aux températures les plus élevées.
Pampuch et al(39) ont proposé un autre mécanisme de réaction à haute température entre le silicium et le carbone. Le carbure de silicium se formerait par précipitation de SiC-β à partir du silicium liquide sursaturé en carbone. La croissance de la couche de carbure de silicium se poursuit à l’interface silicium liquide - carbure de silicium, car la diffusion du carbone dans le carbure de silicium est beaucoup plus rapide que celle du silicium.
4.4.1.3 Défauts d’infiltration réactive
Singh et al(44) ont étudié l’infiltration d’alliages de silicium dans une préforme poreuse de carbone. La microstructure finale est directement liée à la porosité de la préforme poreuse et au cycle d’infiltration. Plusieurs défauts d’infiltration peuvent être mis en évidence, le blocage de l’infiltration, des excès locaux de silicium ou de carbone, des fissures, l’absence de réaction entre le silicium et le carbone.
Le blocage de l’infiltration est essentiellement causé par une porosité trop fine. La réaction entre le carbone et le silicium entraîne une augmentation de volume de 98% par rapport au volume initial de carbone qui peut combler les pores de petite taille et bloquer l’infiltration. La taille des pores qui limitent l’infiltration dépend de l’épaisseur de carbone qui se transforme en carbure de silicium, c’est donc une caractéristique spécifique à chaque système. Cependant, le volume global du mélange silicium carbone diminue d’environ 25% après formation de SiC, laissant de la porosité résiduelle. Les solutions proposées pour palier ces défauts sont l’optimisation de la distribution de la taille des pores ou l’utilisation d’un traitement thermique favorisant l’infiltration par rapport à la réaction(44).
La microstructure du produit final est directement liée à la distribution en taille de la porosité dans la préforme de carbone et aux quantités relatives de silicium et de carbone. Les dispositions pour obtenir une microstructure exempte de silicium ou de carbone libre peuvent être prises en amont ou en aval de l’élaboration. Il faut soit contrôler précisément les conditions d’élaboration, en particulier la distribution de la porosité de la préforme qui doit être resserrée et/ou la quantité de silicium, soit faire subir un post traitement destiné à stabiliser ou éliminer la phase en excès.
4.4.1.4 Les modèles d’infiltration réactive
De nombreux modèles ont été développés pour l’étude de l’infiltration de liquides dans une préforme poreuse. Trois approches sont utilisées. La première tente d’établir une relation entre des caractéristiques microscopiques du matériau et la vitesse d’infiltration, la seconde l’évalue globalement à partir d’une caractéristique macroscopique et la troisième tente d’unifier les deux approches précédentes afin de rendre compte des phénomènes locaux liés à la réaction entre le silicium et le carbone.
Des hypothèses peuvent être formulées dans tous les cas, en particulier si l’épaisseur infiltrée est faible. Les forces de gravité et l’effet de contre pression des gaz emprisonnés dans la porosité sont supposés négligeables face aux forces capillaires(13, 43).
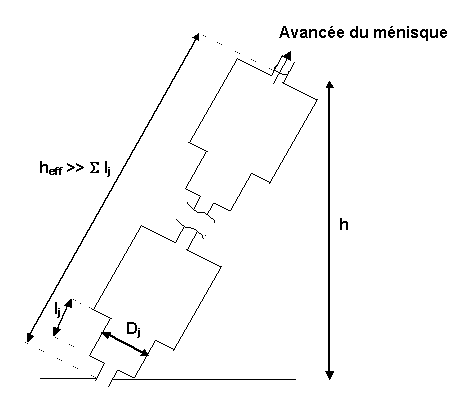
D’un point de vue microscopique, la profondeur d’infiltration peut être déduite de la relation de Hagen-Poiseuille(13, 20, 21, 25, 26) qui exprime la vitesse d’un fluide dans un capillaire en fonction de son rayon r, de sa longueur L et des caractéristiques physiques du fluide :

avec x la profondeur d’infiltration,
t le temps,
γsl la tension de surface du liquide sur le solide,
D le diamètre effectif du capillaire,
θ l’angle de mouillage,
µ la viscosité du fluide.
Plusieurs modèles ont été bâtis pour calculer le diamètre effectif du capillaire en fonction des caractéristiques microscopiques de la préforme.
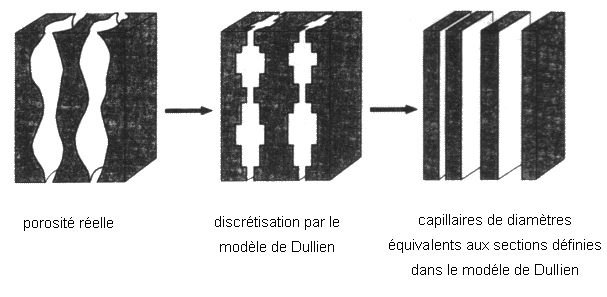
Dullien et al(13) ont considéré l’existence de capillaires parallèles, de diamètres effectifs différents et variables périodiquement (figure 28). Le diamètre effectif (Deff) est exprimé par la relation suivante :
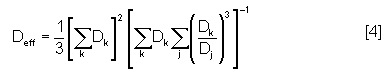
Avec Dj le diamètre d’une section de capillaire de longueur lj (j=1,2….n)
Dk le diamètre d’un capillaire de débit Qk (k=1,2…n)
n le nombre de sections de diamètre constant considérées
Figure 28 : Modélisation de la porosité d’après Dullien(13)
Le diamètre effectif des pores obtenu à partir de cette formule est très inférieur aux diamètres attribués à chaque segment. Ce phénomène peut être expliqué par la résistance à la montée capillaire limitée par les pores de très petit diamètre et par la limitation de la force motrice liée aux pores de grand diamètre, conduisant à une faible vitesse de montée dans le capillaire. Une bonne approximation est donnée en utilisant trois diamètres de pores caractéristiques de la microstructure, celui au point d'inflexion de la courbe v=f(d) obtenue par porosimètrie au mercure, le diamètre maximum déterminé par observation microscopique et la moyenne de ces valeurs.
La représentation de la porosité par un capillaire unique ne semble pas satisfaisante. La porosité est complexe, interconnectée, avec des formes et des tailles de pores différentes. De plus, l’évaluation des caractéristiques physiques du fluide est difficile compte tenu des variations de température associées à la réaction exothermique.
La seconde approche est macroscopique. Elle utilise la loi de Darcy, qui relie la vitesse d’un fluide à travers un matériau poreux à sa perméabilité, grandeur qui permet de considérer le matériau dans son ensemble.

Avec V la vitesse de progression du fluide
K la perméabilité du matériau
ΔP la différence de pression
µ la viscosité du fluide
L la distance d’infiltration
Dans le cadre d’une infiltration spontanée, l’évaluation de ΔP nécessite une approche microscopique du réseau poreux. De plus, la perméabilité est, en général, mesurée en régime stationnaire, alors que l’infiltration est transitoire.
Une expression de la profondeur d’infiltration est donnée par Glass et al(22) :

Avec L la distance d’infiltration,
t le temps,
K la perméabilité,
γsl la tension de surface entre le solide et le liquide,
µ la viscosité du fluide,
θ l’angle de mouillage,
r le diamètre effectif des pores,
p la porosité ouverte totale.
Ils proposent d’utiliser l’expression de la perméabilité déduite du modèle de Carman-Kozeny(6).

Avec K perméabilité
p la porosité totale
k une constante qui prend en compte la forme de la section et la tortuosité des pores
S la surface spécifique par unité de volume
La relation a été validée pour des porosités ouvertes totales comprises entre 30 et 80%(22), mais ce modèle très simple ne reflète ni la complexité de la porosité ni l’influence de la réaction chimique. Une distribution al+éatoire de capillaires de différents diamètres a été introduite dans un réseau à deux dimensions par Fatt(22).
La perméabilité peut aussi être évaluée à partir du modèle de Dullien(13) qui intègre deux grandeurs caractéristiques du matériau, le diamètre moyen des pores obtenu par porosimétrie au mercure et le diamètre du pore le plus grand déterminé par observation au microscope électronique à balayage. La diminution de la taille de la porosité associée à la réaction entre le silicium et le carbone est prise en compte en retranchant l’épaisseur de la couche de SiC formée.
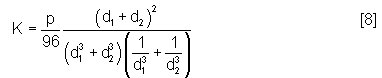
avec K la perméabilité,
d1 le diamètre des pores au point d'inflexion de la courbe de porosimétrie au mercure, après formation de la couche de SiC
d2 le diamètre du pore le plus grand observé au MEB, après formation de la couche de SiC
p la porosité totale
A partir de l’équation de Darcy et du modèle de Dullien(13), Einset et al(14, 15) ont tenté d’introduire l’influence de la réaction chimique sur la perméabilité (équation [9]). Les hypothèses sont basées sur une réaction quasi instantanée au début, formant une couche de SiC d’épaisseur δ et une croissance très lente limitée par un régime diffusionnel de silicium à travers le SiC. Le liquide est supposé parfaitement mouillant (θ=0). Deux diamètres de pores sont utilisés pour la modélisation de la microstructure. Le premier est déterminé au point d’inflexion de la courbe d’intrusion par porosimètrie au mercure (d1) et le second est le pore de diamètre le plus grand observé au MEB (d2). Cependant, à cause de divergences entre le modèle et l’expérience, Einset et al(14, 15) recalculent ce diamètre à partir des mesures de vitesse d’infiltration afin de diminuer cet écart. Une forte dépendance entre δ et d1 et la vitesse d’infiltration a été mise en évidence. Ces deux valeurs sont également corrigées à partir des données expérimentales pour améliorer l’accord entre le modèle et l’expérience.
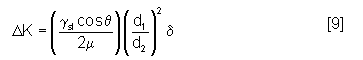
avec ΔK la variation de la perméabilité,
d1 le diamètre du pore au point d’inflexion de la courbe d’intrusion par porosimétrie au mercure,
d2 le diamètre du pore le plus grand,
γsl la tension de surface du fluide sur le solide,
θ l’angle de mouillage du fluide,
µ la viscosité du fluide,
δ l’épaisseur de la couche de SiC formée instantanément.
Le caractère prédictif de ce modèle est très limité car les paramètres les plus influents sont déduits de la mesure de la vitesse d’infiltration.
Sangsuwan et al(43) ont également mis en évidence les insuffisances des modèles basés uniquement sur la loi de Darcy. Un modèle représentatif doit prendre en compte la dynamique des fluides, les interactions thermiques et l’influence de la température sur la siliciuration.
Une troisième approche consiste à unifier les aspects microscopiques et macroscopiques afin d’obtenir une bonne représentation globale de l’infiltration en tenant compte des phénomènes locaux liés à la réaction entre le silicium et le carbone. Gern et al(20, 21) ont affiné la modélisation de Carman-Kozeny(6) en utilisant des capillaires parallèles de diamètres différents en fonction de leur fréquence statistique. L’avancement de l’infiltration est évalué à partir de la concentration en silicium dans la préforme. La loi de Fick permet de calculer la vitesse d’infiltration à partir du coefficient de « diffusion » D introduit par Hillig et al(25, 26).

Avec r le rayon effectif du capillaire dans le cas d’un gaz
r0 le rayon réel du capillaire
R le rayon effectif du capillaire dans le cas d’un fluide
f la tortuosité de la porosité
γ la tension de surface du fluide
η la viscosité du fluide
Figure 29 : Modélisation du système pour le calcul de la concentration en silicum d’après Gern et al(20, 21)
Reed et al(41) ont montré que la perméabilité n’est pas bien décrite à partir du modèle de Carman-Kozeny mais qu’elle pouvait l’être en utilisant la porosité totale et la distribution de la taille des pores, déterminée par porosimètrie au mercure. Ainsi, pour les compacts de particules, la perméabilité peut être obtenue par la théorie de la percolation avec :

avec K la perméabilité
p la porosité totale
r la fonction de distribution de la taille des pores
c une constante empirique
A chaque taille de pore est associée une conductance. Il existe une conductance limite en deçà de laquelle le fluide ne passe pas. Aucune expérience n’est venue confirmer ou infirmer cette hypothèse.
Rajesh et al(40) ont associé les modèles macroscopique et microscopique afin de modéliser l’infiltration en tenant compte de la réaction exothermique entre le silicium et le carbone d’une préforme de fibres. La perméabilité est évaluée à partir d’un élément représentatif de la microstructure (figure 30) décrit d’après l’approche de Watson. L’objectif est d’évaluer et de modéliser l’évolution de la porosité en fonction de la réaction entre le silicium et le carbone.
Les limitations des modèles sont essentiellement dues au choix de la forme des pores et de la représentation de la distribution en taille des pores ou de sa variation au cours du temps, dans le cadre d’un phénomène d’infiltration réactive.
La perméabilité diminue rapidement en deçà d’une certaine taille de pores car l’interconnexion des pores chute et la tortuosité augmente. Un autre problème vient de l’emprisonnement du gaz contenu dans la porosité, quand le liquide s’infiltre de tous les côtés. La valeur de perméabilité déterminée en régime stationnaire est alors, largement surévaluée. L’approche utilisée par Rajesh et al(40) semble mener à des résultats intéressants, en modélisant des phénomènes microscopiques et macroscopiques. Cependant, la transposition à notre système est difficile compte tenu de la différence entre les porosités d’une préforme de fibres et d’un compact de poudre.
Figure 30 : Elément volumique représentatif de la microstructure d’un tissu de fibres d’après Watson(40)
4.4.2 Protocole expérimental
La suspension de silicium est préparée à partir de poudre de silicium et de carboxyméthylcellulose (CMC) et d’eau. L’ensemble est désaggloméré et homogénéisé aux ultrasons.
Les substrats revêtus de HfB2 et de carbone sont enduits par immersion dans cette suspension, puis étuvés afin d’éliminer l’essentiel de l’eau introduite.
La masse des échantillons est mesurée après chaque étape afin d’évaluer les quantités de mélange HfB2+C et de silicium apportées par les enductions et pour déterminer la masse du revêtement final.
Le traitement thermique est réalisé, sous atmosphère inerte. La température est mesurée par un pyromètre bichromatique qui permet de s’affranchir de la nature du matériau sur lequel la visée est réalisée.
La vitesse de montée en température est de 16.67.10-2°/s, jusqu’à la température de palier. La durée de ce dernier est variable et la descente en température est contrôlée par l’inertie du four.
Les phases formées sont déterminées par diffraction des rayons X (Siemens D5000), analyse chimique, observation au MET (JEOL 2010) de lames minces et par spectrométrie Auger (VG microlab 310F). La microstructure des monolithes est caractérisée quantitativement par la porosimétrie au mercure (micromeritics autopore II 9220) et la surface spécifique BET (micromeritics ASAP 2010). Des observations systématiques de sections polies au MEB (Philips XL30) et par microsonde X (Cameca SX100) sont menées sur les deux types d’échantillons.
4.4.3 Influence des paramètres de siliciuration
Dans un premier temps, nous avons cherché à définir les conditions expérimentales à partir des modèles développés précédemment.
Nous avons tenté d’évaluer le temps nécessaire pour infiltrer des monolithes HfB2+C de 200.10-6 m d’épaisseur, sous argon, à 1703K, qui est la température la plus basse, mesurée dans le four, pour laquelle le silicium est totalement fondu.
Les forces de gravité et la contre pression exercée par l’argon piégé dans la porosité sont négligées par rapport aux forces capillaires, compte tenu de la faible épaisseur infiltrée et de la grande porosité du substrat. L’angle de mouillage du silicium est variable. A sa fusion, il est probablement proche de 0° et augmente pour atteindre une valeur supérieure à 90°. L’équilibre est atteint en quelques secondes. Nous avons donc évalué le temps d’infiltration dans deux cas extrêmes, pour des angles de mouillage de 10° et de 80°, car il doit être inférieur à 90° pour que l’infiltration soit spontanée. L’influence de la température ne sera pas introduite par manque de données expérimentales. Nous pouvons seulement dire qu’elle est supérieure à 1703K à cause du caractère exothermique de la réaction.
Deux hypothèses peuvent être envisagées, soit l’infiltration est plus rapide que la réaction, soit la réaction est quasi instantanée par rapport à l’infiltration.
La modélisation de la microstructure développée par Dullien et al(13) permet de calculer le temps d’infiltration à partir de l’équation [3] et des données du tableau 9.
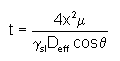
Avec t le temps en seconde
x la profondeur d’infiltration
µ la viscosité du silicium à sa température de fusion
γsl la tension de surface du silicium liquide
θ l’angle de mouillage du silicium sur le carbone
Dapp le diamètre apparent de la porosité calculé à partir de la relation de Dullien et al(13) (équation [4])
La première hypothèse suppose que la réaction est lente par rapport à l’infiltration. Le diamètre apparent doit être calculé à partir des diamètres déterminés par porosimétrie au mercure sur les échantillons pyrolysés.
Pour la seconde hypothèse la réaction est quasi instantanée. Il se forme une couche de SiC dont l’épaisseur peut être évaluée à partir de la quantité de carbone dans la préforme poreuse et la surface spécifique de la poudre de HfB2, en supposant que la résine se répartisse uniformément à la surface de la poudre (tableau 10).
Tableau 11 : Calculs du temps d’infiltration dans les deux hypothèses

En première approximation, le temps nécessaire à l’infiltration d’une épaisseur de 200.10-6 m de mélange HfB2+C est de l’ordre du centième de seconde dans le plus défavorable des cas. L’infiltration ne semble donc pas être l’étape limitante du procédé d’élaboration. La formation de la couche d’accrochage à l’interface et la réaction entre le silicium liquide et le carbone nécessitent plus de temps pour obtenir une microstructure homogène. La durée du palier sera donc fixée à 3600 s afin de favoriser ces réactions.
4.4.3.1 L’atmosphère de siliciuration
L’atmosphère qui doit être non oxydante (inerte ou vide), afin de protéger le substrat de toute oxydation pendant le traitement, joue un rôle important dans les phénomènes de mouillage. Il est donc important d’évaluer le comportement du silicium liquide dans les atmosphères retenues.
Les propriétés physiques du silicium liquide, en particulier sa mouillabilité dépendent également de la nature de la surface du substrat et de la teneur en oxygène introduite lors du stockage à l’air et de sa mise en suspension dans l’eau. L’effet de contre pression exercée par le gaz emprisonné dans la porosité peut être éliminé par un traitement sous vide.
Compte tenu de la difficulté d’extrapoler les résultats de mouillage, publiés dans la littérature, à nos conditions expérimentales, une évaluation a été réalisée sur du graphite siliciuré pour représenter le carbure de silicium qui se forme quasi instantanément pendant l’infiltration réactive. La figure 31 montre que le silicium mouille parfaitement sous vide dynamique primaire (a), alors qu’il est non mouillant sous argon (b). On observe des billes de silicium et de la poudre de carbure de silicium mais le tout est facilement éliminé par brossage (partie inférieure de la photo b).
Figure 31 : Test de mouillabilité du silicium sur du graphite siliciuré, sous vide primaire dynamique a) et sous argon b).

Les microstructures des revêtements siliciurés à 1703K pendant 3600s sont présentées sur les figures 32 à 34. Nous obtenons les deux comportements observés lors de l’étude préliminaire mais la microstructure des revêtements est reproductible, ce qui montre l’importance de l’étape de broyage et du contôle de la distribution de la porosité de la préforme poreuse.
Les revêtements obtenus sous vide sont caractérisés par une couche externe dense, riche en silicium libre et solidaire du revêtement qui est compact. On note un fort accrochage et un grossissement des grains de HfB2 (figure 33).
Le vide primaire favorise un bon mouillage du silicium, l’élimination de l’effet de contre pression, et par conséquent l’infiltration. On retrouve du silicium en profondeur dans la porosité du substrat. Cependant, dans ces conditions de traitement, le contrôle de la quantité de silicium apportée lors de l’enduction semble être très important pour limiter sa présence en surface.
Figure 32 : Microstructure obtenue par siliciuration sous vide dynamique primaire, à 1703K/3600s

Figure 33 : Micrographie au MEB du grossissement des grains de HfB2 après siliciuration sous vide dynamique primaire

Figure 34 : Microstructure obtenue par siliciuration sous argon, à 1703K/3600s.

Les revêtements élaborés sous argon sont recouverts d’une couche de poudre de carbure de silicium et de billes de silicium faciles à éliminer par brossage (figure 34). La microstructure est plus homogène et plus fine que sous vide. La morphologie des grains de HfB2 semble indiquer que les processus de dissolution-reprécipitation avec grossissement des grains sont moins importants. Ce phénomène peut être attribué à un volume infiltré de silicium liquide plus faible en raison de sa mauvaise mouillabilité. Cependant, l’infiltration semble complète puisque l’on retrouve du carbure de silicium à l’interface entre le substrat et le revêtement et que ce dernier semble dense (figure 35).
Les forces capillaires sont suffisamment élevées pour que le silicium atteigne l’interface avec le substrat. Cependant, une grande partie du silicium en excès se retrouve sous forme de billes, ce qui indique une faible mouillabilité. L’utilisation de l’argon présente donc l’intérêt considérable de ne pas nécessiter un contrôle local précis de la quantité de silicium apportée lors de l’enduction, ce qui est compatible avec la technique d’enduction utilisée.
Les deux atmosphères testées présentent des avantages et des inconvénients. Sous vide l’infiltration est excellente mais la quantité de silicium résiduelle est très importante et les grains subissent un fort grossissement. Sous argon, l’infiltration semble complète, la majeure partie du silicium en excès peut être éliminée de manière simple et la taille des grains évolue peu.
Nous avons donc modifié les conditions expérimentales pour concilier les avantages apportés par chaque atmosphère. L’infiltration débute par un traitement sous vide, pour favoriser l’infiltration, puis est poursuivie sous argon après 1800s de palier à 1703K, pour éliminer le silicium en excès. Cependant, les observations de sections polies au microscope électronique à balayage montrent que la microstructure obtenue est identique à celle formée sous vide.
Figure 35 : Cartographie par microsonde X d’une section polie de revêtement siliciuré sous argon à 1703K pendant 3600s (interface substrat / revêtement)

4.4.3.2 Température et temps de palier
La température de traitement doit être la plus faible possible afin de limiter la fissuration du revêtement due aux contraintes thermiques entre le revêtement et le substrat lors du retour à température ambiante. La cinétique de formation du carbure de silicium doit être rapide compte tenu de la faible épaisseur de carbone et de sa forte réactivité, si la microstructure est parfaitement homogène. De plus, la réaction entre le silicium et le carbone est fortement exothermique.
Les caractéristiques physico-chimiques du silicium liquide, les vitesses des réactions chimiques et les contraintes mécaniques résiduelles, dépendent de la température. Il faut donc trouver un compromis entre une température élevée, qui favorise le mouillage et accélère les cinétiques de réaction et une température faible qui limite les contraintes thermiques, la température la plus faible possible étant la température de fusion du silicium. Compte tenu de la rapidité de l’infiltration, la durée du palier agit essentiellement sur l’avancement des réactions chimiques et le grossissement des grains. Nous avons réalisé divers traitements thermiques sous argon dont les températures et les durées des paliers sont consignées dans le tableau 12.
| Essai | 1er palier |
2eme palier |
||
| T (K) | Durée (s) | T (K) | Durée (s) | |
| 1 | 1703 | 3600 | - | - |
| 2 | 1703 | 7200 | - | - |
| 3 | 1703 | 3600 | 1873 | 3600 |
| 4 | 1703 | 3600 | 1873 | 7200 |
Les observations de sections polies confirment une infiltration du silicium jusqu’à l’interface substrat/revêtement quelles que soient les conditions expérimentales. L’allongement de la durée du palier à 1703K ne semble pas modifier la microstructure. Cependant après traitement à 1873K l’élimination par brossage de la poudre de carbure de silicium et des billes se silicium superficielles est difficile à cause d’un début de « densification ». De plus, des fissures sont apparues sur les échantillons ayant subi un traitement à 1873K pendant 3600s. Ainsi, afin d’obtenir des résultats comparables, nous avons choisi de conserver la température de 1703K et une durée de 3600s dans tous les traitements de siliciuration.
Un post traitement peut être envisagé, mais uniquement après élimination de la couche de poudre de carbure de silicium et des billes de silicium dans le cadre d’une siliciuration sous argon.
4.4.3.3 Composition de la suspension de silicium
Une quantité trop faible de silicium apportée par l’enduction peut engendrer des défauts d’infiltration. Nous avons donc étudié son influence sur la microstructure du revêtement après traitement thermique sous vide ou sous argon (figure 36).
Quatre suspensions avec des teneurs différentes en silicium ont été préparées et les enductions réalisées selon le même protocole (tableau 13). Des sections polies sont observées systématiquement au MEB (figure 37, figure 38).
La quantité de silicium nécessaire à la siliciuration de la préforme HfB2+C est calculée à partir de la masse de revêtement en prenant un rendement en carbone de 35% pour la résine. Le rapport de la masse de silicium déposée par l’enduction sur la masse théorique pour obtenir SiC est présenté dans le tableau 21. Les moyennes sont effectuées sur trois échantillons.
Des différences de comportement sont visibles à la sortie du four des échantillons (figure 36). Sous vide, la quantité de silicium libre en surface diminue lorsque le rapport décroît, ce qui confirme la nécessité d’apporter la quantité exacte de silicium lors de l’enduction. Sous argon, il se forme de la poudre de carbure de silicium dans tous les cas, mais la taille et le nombre des billes de silicium diminuent avec une quantité de silicium décroissante, pour disparaître à partir d’un léger excès de silicium.
Des défauts d’infiltration apparaissent sous vide avec une porosité localisée importante. Les microstructures obtenues sous vide et pour des teneurs élevées en silicium mettent en évidence un grossissement des grains de HfB2 (figure 37a et 37b). Pour tous les revêtements on ne décèle par de front d’infiltration du silicium.
Sous argon les microstructures obtenues sont très proches quelle que soit la composition de la suspension. Les grains ont des tailles comparables à la distribution de la poudre initiale et l’infiltration est complète dans toute l’épaisseur.
Ces observations de sections polies mettent en évidence une influence plus marquée de la teneur en silicium sur le développement de la microstructure pour une siliciuration sous vide (figure 37) que sous argon (figure 38).
Figure 36 : Aspects des échantillons à la sortie du four en fonction de la quantité de silicium et de l’atmosphère de traitement
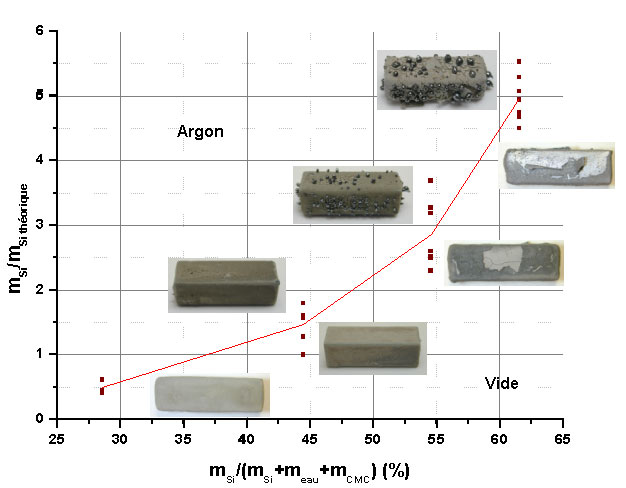
Figure 37 : Evolution de la microstructure de sections polies de revêtements élaborés sous vide en fonction de la quantité de silicium dans la suspension

Figure 38 : Evolution de la microstructure de sections polies de revêtements élaborés sous argon en fonction de la quantité de silicium dans la suspension

4.4.4 Discussion
Les revêtements obtenus par infiltration sous vide et sous argon ont des caractéristiques différentes. La succession des différentes étapes de la siliciuration va être discutée en fonction de l’atmosphère.
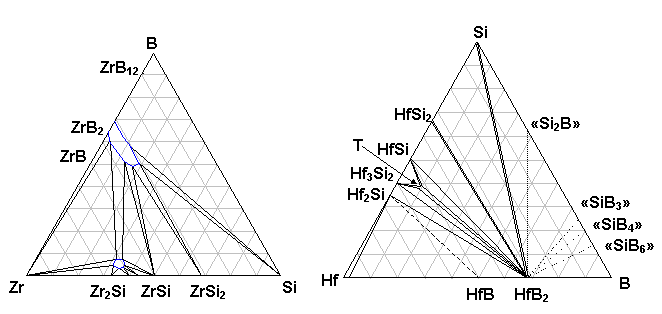
La montée en température s’effectuant sous vide, dès que le domaine1523-1573K est atteint une partie de l’oxygène présent à la surface des grains de poudre de silicium s’élimine sous forme de SiO, puis après fusion (1693-1703K) le silicium nappe toute la surface de l’échantillon. Sous vide primaire le silicium liquide mouille parfaitement les grains de HfB2 recouverts de carbone et s’infiltre dans la porosité jusqu’au substrat. Les calculs du temps d’infiltration montrent qu’il est très court. La durée nécessaire pour atteindre l’équilibre pour le mouillage du carbone par le silicium est de l’ordre de quelques secondes, ce qui est largement suffisant pour permettre son infiltration. Un large volume de la porosité contient du silicium tout au long du palier, ce qui entraîne un grossissement des grains de HfB2, probablement par un phénomène de dissolution reprécipitation. Par analogie avec le système ZrB2-Si (figure 39) on peut en déduire l’existence d’un eutectique dans le système HfB2-Si à plus haute température. Mais il a été montré que la température atteinte dans le préforme infiltrée est supérieure à celle imposée par le cycle à cause du caractère exothermique de la réaction entre le carbone et le silicium (18, 16). Compte tenu de la forte adhésion du silicium sur le carbure formé, l’excès de silicium resté à la périphérie de l’échantillon ne peut pas être éliminé de manière simple. Un post traitement thermique est nécessaire.
Les phénomènes mis en jeu lors du traitement sous argon semblent plus complexes en raison de la présence de gaz dans la porosité qui peut freiner la progression du silicium liquide. Le mauvais mouillage du silicium peut être dû à la présence d’oxygène à la surface des grains. A l’instant initial, il y a une compétition entre l’infiltration du silicium et la formation de gouttes entourées par un film d’oxyde ou de carbure. Sous l’action des forces capillaires le silicium progresse jusqu’à ce que la contre pression gazeuse bloque la progression ou que l’interface avec le substrat soit atteinte. La poudre de carbure de silicium retrouvée à la surface de l’échantillon serait le produit d’une réaction entre le carbone et les vapeurs de silicium tout au long du palier de traitement.
Les observations microscopiques et les analyses montrent que l’infiltration réactive du silicium sous argon à travers la préforme HfB2+C est complète. Elle présente l’avantage par rapport au vide de permettre l’élimination facile du silicium en excès, ce qui évite un contrôle rigoureux de la quantité de silicium apportée par l’enduction. La microstructure obtenue est dense, avec une faible quantité de silicium libre. L’augmentation de la température ou de la durée du palier n’a pas montré d’effet bénéfique.
Figure 39 : Diagrammes de phase des systèmes Zr-B-Si à 1623K(30) et Hf-B-Si à 1573K, T est une phase inconnue (5).
4.4.5 Conclusions
L’infiltration réactive sous argon permet de mieux contrôler la quantité de silicium incorporée dans le revêtement. Dans les deux cas, l’infiltration de la préforme HfB2+C est complète et il se forme une couche d’accrochage en carbure de silicium à l’interface. La majeure partie du silicium en excès s’élimine très facilement après traitement sous argon, contrairement au vide. Pour ces raisons, le cycle de siliciuration choisi est un palier de 3600s à 1703K, sous argon, après une montée en température à 16.67.10-2°/s. Un post traitement peut être nécessaire en fonction de l’application visée, par exemple pour éliminer le silicium en excès.
4.5 Analyse microstructurale
A haute température, le comportement à l’oxydation d’un matériau est étroitement lié à la porosité ouverte totale, à sa distribution en taille et à la répartition des phases. Nous avons cherché à caractériser l’évolution de la microstructure entre l’état pyrolysé et l’état siliciuré pour avoir des indications sur la qualité de l’infiltration réactive et sur les conditions expérimentales d’élaboration les mieux adaptées.
La composition chimique a été mesurée aux différentes étapes du procédé d’élaboration afin de contrôler chaque traitement et d’y apporter des mesures correctives. Des difficultés de mise en solution du silicium et de l’hafnium ont entraîné quelques imprécisions sur la détermination de leurs teneurs et la multiplication des essais.
Les analyses chimiques présentées dans le tableau 14 font apparaître une bonne reproductibilité des compositions après pyrolyse. Nous constatons cependant des divergences après siliciuration, sur ces résultats bruts, avec un rapport Si/C qui varie de 1,34 à 2,36. Il semble difficile d’attribuer de tels écarts aux variations de la distribution de la taille des pores.
La distribution en taille de la porosité ouverte (figure 40) montre que l’infiltration réactive est assez reproductible. L’intrusion du silicium s’effectue essentiellement dans les pores de diamètre proche de 10-6 m. Après siliciuration, la taille moyenne des pores est comprise entre 0,09.10-6 et 0,16.10-6 m, avec une distribution assez large. Malgré des surfaces spécifiques voisines et des distributions de la taille des pores assez proches à l’état pyrolysé, des écarts apparaissent à l’issue de la siliciuration. Cependant, ces mesures confirment la bonne infiltration du silicium puisque la surface spécifique diminue largement (tableau 15).
La porosité ouverte totale, mesurée par poussée d’Archimède, sur des monolithes siliciurés est inférieure à 10%.
La figure 41 montre l’observation au MEB d’une section polie d’un revêtement élaboré sur du graphite et siliciuré sous argon.
Tableau 15: Mesure de surface spécifique au Krypton sur monolithes pyrolysés et siliciurés sous argon

Figure 40 : Distribution de la taille des pores après infiltration réactive
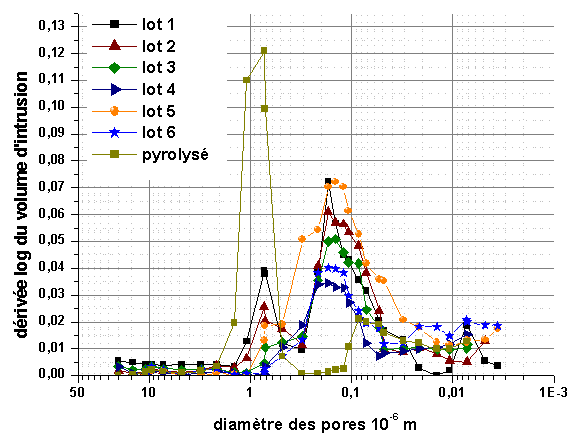
Figure 41 : Observation au microscope électronique à balayage d’une section polie d’un revêtement siliciuré sous argon

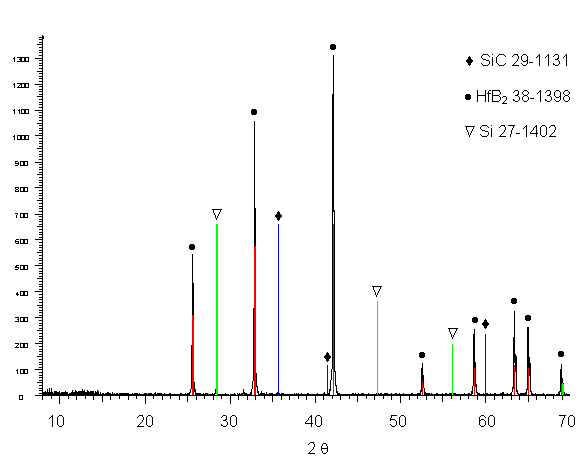
Les spectres de diffraction des rayons X obtenus sur des monolithes siliciurés sous argon, broyés au mortier, montrent la présence d’une faible quantité de silicium libre et une faible intensité de diffraction du carbure de silicium (figure 42).
Une analyse radiocristallographique plus précise avec un balayage plus lent a été réalisée. Aucune modification du spectre de diffraction de HfB2 n’a été observée et les raies correspondant au carbure de silicium restent de faible intensité malgré sa teneur dans le matériau. En réalité, comme le montre l’étude en MET présentée ultérieurement, la majorité des grains sont de très petite taille et ne sont donc pas détectés par les rayons X.
Figure 42 : Diagramme de diffraction des rayons X de monolithes broyés, siliciurés sous argon
Des analyses par spectrométrie Auger ont été entreprises afin de localiser et de déterminer la nature des différentes phases. Les échantillons sont polis soigneusement et la surface « nettoyée » avant chaque analyse. Un exemple représentatif des cartographies des éléments est reproduit sur la figure 43. Les micrographies confirment la distribution homogène de HfB2. On observe le frittage d’agglomérats, mais le grossissement des grains reste faible. Pour le carbone et le silicium, il existe des régions où les deux éléments sont présents, ce qui met en évidence la formation de SiC. En revanche, on observe des domaines où le silicium est largement excédentaire, qui doivent probablement correspondre à des volumes poreux importants avant l’étape de siliciuration. Des profils de composition ont été réalisés sur un grand nombre de grains afin d’analyser l’interface HfB2/matrice SiC (figure 44). Les courbes donnant la teneur d’un élément en fonction de la distance font apparaître des gradients de composition en périphérie de tous les grains de HfB2.
Bien qu’il n’existe pas de diagramme de phase pour le système Hf/B/Si/C, il est possible d’envisager l’existence d’un eutectique à la température de siliciuration. La présence de cet eutectique expliquerait l’évolution de la taille des grains de HfB2 par dissolution des petits grains, migration des espèces dans le liquide et reprécipitation sur les grains de fort diamètre. Le phénomène de reprécipitation pourrait expliquer la présence de gradients de composition.
Dans le but d’obtenir des informations supplémentaires sur les domaines contenant le carbure de silicium et l’interface HfB2/matrice une étude en microscopie électronique en transmission a été entreprise. Les échantillons ont été coupés à partir de monolithes, polis et amincis par bombardement ionique. Les observations mettent en évidence des grains de HfB2 entourés par une matrice mal organisée. Les anneaux de diffraction observés sur le cliché de la figure 45 sont dus à la taille nanométrique des cristaux qui constituent la matrice.
Compte tenu de la différence de dureté entre les grains de HfB2 et la matrice de SiC, il n’a pas été possible d’obtenir de grains suffisamment minces pour analyser le gradient de composition.
Figure 43 : Cartographies du silicium, du carbone et de l’hafnium par spectrométrie Auger

Figure 44 : Analyse auger de l’interface entre un grain de HfB2 et la matrice de carbure de silicium suivant la ligne L2 (photo)
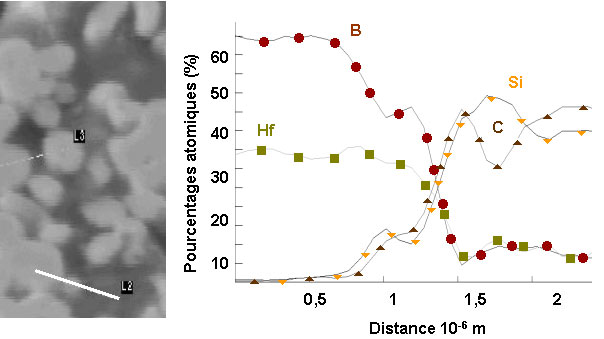
Figure 45 : Cliché MET d’une lame mince de monolithe après siliciuration sous argon à 1703K pendant 3600s

4.6 Discussion et conclusions
Les mesures de porosimètrie par intrusion de mercure et de surface spécifique mettent en évidence une infiltration complète de la préforme HfB2+C. L’homogénéité de la répartition du silicium semble directement liée à la distribution en taille des pores de la préforme. Les variations locales entraînent la formation de quelques zones riches en silicium ou en carbone libre. L’ensemble des résultats montre qu’une distribution en taille étroite est nécessaire pour obtenir un matériau homogène. La porosité développée est essentiellement contrôlée par l’empilement des grains de HfB2, la résine contribuant à la formation d’une porosité très fine mais de grande surface spécifique, favorable à une bonne réaction chimique entre le silicium et le carbone.
Deux phases sont issues de l’infiltration du silicium. La première par réaction chimique entre le silicium et le carbone. Elle conduit à la formation de nanoparticules de carbure de silicium, probablement en raison de la taille nanométrique du carbone issu de la résine phénolique(42) et de la température de siliciuration qui ne favorise pas le frittage de SiC. La seconde phase formée est localisée à l’interface entre les grains de HfB2 et le mélange silicium, carbure de silicium. Il existe probablement un eutectique dans le système Hf-B-Si-C dont la température de formation est proche de celle du palier de siliciuration. Cependant, le caractère exothermique de la réaction entre le silicium et le carbone peut permettre des augmentations locales de température, en particulier à la périphérie des grains de HfB2, zone de forte concentration en carbone. Ce phénomène transitoire a été observé par Einset et al(15) lors de l’infiltration de carbone poreux. A la fin de la réaction, la température diminue pour revenir à la température de palier(14, 43), ce qui conduit à la précipitation de l’eutectique à la surface des grains de HfB2 sur une épaisseur assez faible d’environ 0,5.10-6 m.
5. TRAITEMENT POST SILICIURATION
Suivant l’application visée, certaines phases nécessitent d’être stabilisées ou éliminées. Les analyses par diffraction des rayons X et la spectrométrie Auger montrent la présence de silicium libre résiduel. Deux types de traitements thermiques post siliciuration ont été testés pour éliminer le silicium excédentaire.
5.1 Post traitement sous argon
L’objectif de ce traitement est d’éliminer le silicium libre par évaporation. Seuls les échantillons siliciurés sous argon ont été traités compte tenu de la faible proportion de silicium libre par rapport aux échantillons siliciurés sous vide.
Les effets du traitement sur la composition globale ont été évalués par analyse thermogravimétrique. L’évolution de la microstructure a été suivie par mesure de la surface spécifique et de la porosité ouverte.
Le traitement est réalisé en montée linéaire sous argon jusqu’à 1973K puis la température est maintenue pendant 7200s. L’analyse thermogravimétrique montre une perte de masse très rapide à partir de 1673K qui se ralentit progressivement après 3600s pour atteindre pratiquement une asymptote. La perte de masse est de l’ordre de 2%.
La distribution de la taille des pores s’élargit et la taille moyenne augmente (figure 46). Cette évolution est confirmée par les mesures de surface spécifique (tableau 16). Ces résultats sont en accord avec les analyses Auger qui montrent que le silicium libre se situe préférentiellement dans les pores de grande dimension (figure 43).
| Après siliciuration | Après traitement sous argon à 1973K/7200s | |
| Surface spécifique en m²/kg | 600 | 1400 |
Figure 46 : Evolution de la porosité aux différentes étapes de traitement
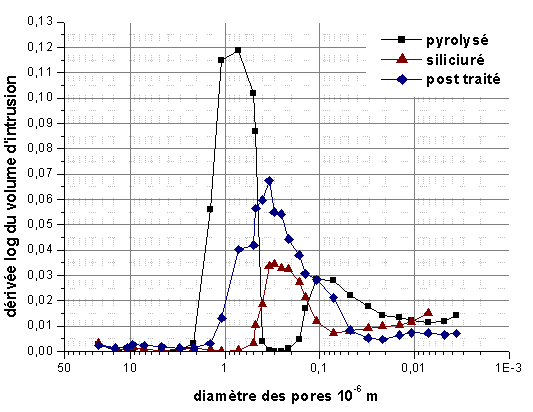
En conclusion, cette expérimentation montre qu’un traitement thermique sous argon conduit à une évolution microstructurale trop importante qui est incompatible avec l’objectif visé d’une meilleure tenue du matériau à haute température.
5.2 Post traitement sous azote
L’objectif de ce traitement est de diminuer la teneur en silicium libre et de profiter de l’augmentation de volume lors de la réaction de nitruration pour éviter l’accroissement de la porosité ouverte observée lors du chauffage sous argon.
Un compromis entre vitesse de réaction et évaporation du silicium doit être trouvé. Nous avons choisi d’effectuer un palier à 1823K avec un ralentissement de la vitesse de montée à partir de 1473K, température à laquelle la réaction avec l’azote débute.
L’analyse thermogravimètrique du traitement sous azote (figure 47) montre deux gains de masse rapides à 1573K et à 1823K mais la réaction particulièrement lente s’échelonne sur une douzaine d’heures. A l’issue du traitement, des petites billes de silicium apparaissent à la surface de l’échantillon. Les analyses chimiques mettent en évidence l’incorporation d’une importante quantité d’azote et la diminution de la teneur en silicium (tableau 17). De plus, l’effet sur la distribution de la taille des pores est très faible comparé à celui observé après traitement sous argon (figure 48).
Figure 47 : Analyse thermogravimètrique du traitement sous azote
Tableau 17 : Evolution de la composition du matériau après traitement sous azote

Figure 48 : Evolution de la porosité aux différentes étapes de traitement
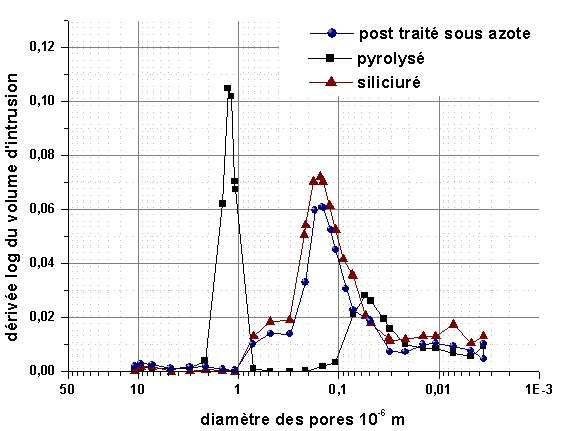
La localisation de l’azote incorporé lors du traitement thermique a été réalisée en déterminant les phases formées, par diffraction des rayons X, et par spectrométrie Auger.
Le diagramme de diffraction des rayons X met en évidence la formation d’une phase intermédiaire de type HfN1-xB2x. Cette phase non stœchiométrique est mentionnée dans le diagramme ternaire Hf-B-N proposé par Smid et al(45). Cependant la réaction de nitruration à 1823K de HfB2 en présence de silicium libre devrait se traduire par la formation de nitrure de silicium ou de bore. Or ces composés ne sont pas détectés. De plus, la quantité extrêmement faible, de silicium libre résiduel peut être associée à la présence de quelques billes formées à la surface des échantillons. Les intensités des pics de diffraction du carbure de silicium sont toujours faibles, indiquant que le traitement a eu peu d’influence sur le grossissement des cristaux.
Figure 49 : Diagramme de diffraction des rayons X d’un monolithe broyé, post traité sous azote à 1823K pendant 14400s.
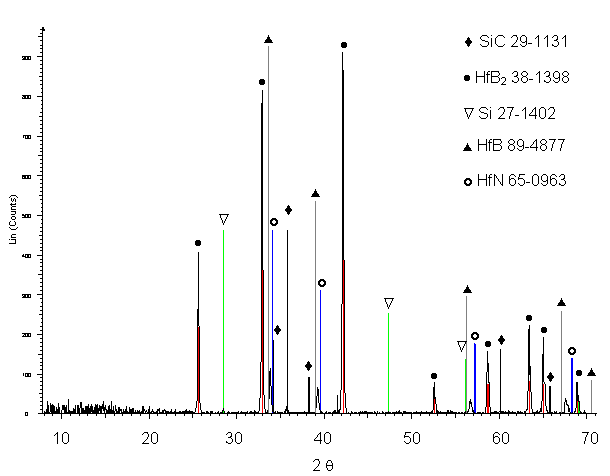
Les cartographies de Hf, N et Hf+B, réalisées par spectrométrie Auger, montrent que certains grains de borure contiennent de l’azote (figure 50). Les zones gris clair sur la cartographie de Hf+B correspondent au composé intermédiaire HfN1-xB2x détecté par diffraction des rayons X. Elles sont cependant en nombre restreint par rapport à HfB2.
Figure 50 : Cartographies de l’hafnium, de l’hafnium + bore et de l’azote par spectrométrie Auger
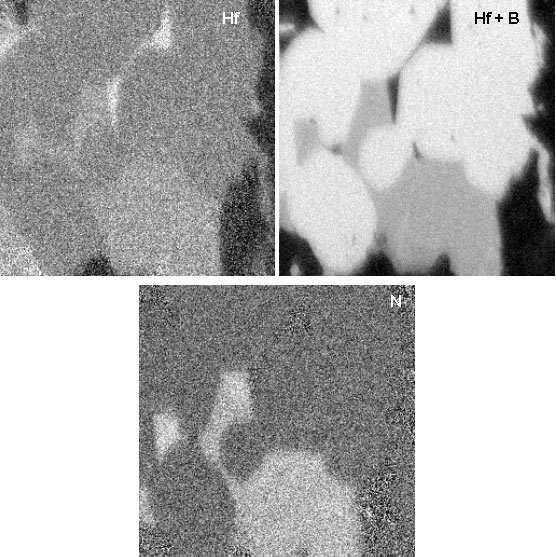
Le traitement de nitruration permet d’éliminer la majeure partie du silicium libre avec une variation très faible de la porosité. Cependant, il est difficile d’avoir accès à la composition minérale globale des matériaux élaborés compte tenu de leur microstructure, des gradients de composition à la périphérie des grains de HfB2 et de la présence d’un boronitrure de hafnium, comme le montrent les analyses par spectrométrie Auger.
6 ELABORATION DE REVETEMENTS
De nombreux matériaux composites à base de carbone et de carbure de silicium existent ou sont en cours de développement dans l’industrie spatiale. La possibilité de les protéger contre l’oxydation permet d’élargir leurs champs d’utilisation. Pour cette raison, le procédé d’élaboration que nous avons mis au point a été testé sur des composites d’architectures et de compositions variées.
La transposition du procédé aux composites C/SiC et C/C siliciurés peut induire des problèmes d’adhésion liés à la présence de carbure de silicium qui pourrait limiter la formation de la couche d’accrochage et l’infiltration du silicium dans la porosité du composite.
De plus, les composites d’architecture complexes présentent une rugosité de surface importante et des différences de mouillabilité en fonction de l’orientation des fibres de carbone ce qui peut générer des variations de l’épaisseur du revêtement.
6.1 Protocole expérimental
Les substrats sont usinés pour éliminer toutes les arêtes vives, sources de contraintes et d’écaillage. Ils sont ensuite nettoyés aux ultrasons dans l’éthanol afin d’éliminer la poussière de carbone en surface et séchés en étuve.
Le procédé d’élaboration est celui présenté et mis au point dans les chapitres précédents.
6.2 Résultats
L’enduction et l’infiltration du silicium sont bonnes quelle que soit l’architecture du composite (figure 51, 52, 53, 54). Un accrochage chimique et mécanique est obtenu par infiltration et réaction entre le silicium liquide et les substrats à base de carbone, comme le montrent les micrographies. Les composites carbone/SiC peuvent présenter deux difficultés, la matrice de carbure peut réagir faiblement avec le silicium et la présence d’une couche de finition en SiC diminue la porosité ouverte du substrat. En réalité, les revêtements élaborés ont une bonne adhérence. Cependant, aucune mesure quantitative n’ayant été effectuée, il est difficile d’évaluer l’impact de la nature du composite sur l’adhésion.
La texture de la surface et l’orientation des fibres peuvent influencer localement l’épaisseur déposée lors de l’enduction. Une légère diminution est mesurée au sommet des boucles de fibres de carbone présentent sur une face du composite à fibres continues (figure 54). Des variations d’épaisseur existent également sur les composites à fibres non continues (figure 51), probablement à cause de la différence de mouillabilité entre les surfaces constituées des extrémités des fibres et celles présentant les faces latérales des fibres. Cependant, ces variations restent limitées à environ 15%.
Ainsi, le procédé développé permet d’élaborer un revêtement HfB2-SiC sur tous les types de composites testés, indépendamment de leur architecture, de l’orientation des fibres à la surface et de la présence d’une couche de protection ou d’une matrice différente du carbone. Des mesures quantitatives seraient nécessaires pour évaluer l’impact de ces différents paramètres sur les performances de la protection.
Figure 51 : Section polie d’un revêtement élaboré sur composite 2,5D C/C

Figure 52 : Section polie d’un revêtement élaboré sur composite C/C siliciuré

Figure 53 : Section polie d’un revêtement élaboré sur composite 2,5D C/SiC fils méchés

Figure 54 : Section polie d’un revêtement élaboré sur composite 3D C/C fils continus

7. CONCLUSIONS
Un nouveau procédé d’élaboration de revêtement HfB2-SiC sur des substrats carbonés par infiltration réactive de silicium liquide dans une matrice de HfB2+C a été développé.
La porosité du mélange HfB2+C, issu de la décomposition thermique d’une résine phénolique, détermine l’homogénéité de la répartition de HfB2, la qualité de l’infiltration du silicium et la présence éventuelle de défauts. L’optimisation des caractéristiques de la suspension de HfB2 et en particulier la granulométrie de la poudre de HfB2 a permis d’obtenir des revêtements avec une porosité homogène et étroitement distribuée autour de 10-6 m. Cette microstructure s’est avérée compatible avec les caractéristiques requises pour l’infiltration du silicium à 1703K, sous argon.
Cette étape a fait l’objet d’une étude particulière. Les phénomènes mis en jeu sont nombreux et complexes à maîtriser. Un revêtement compact et homogène constitué de grains de HfB2 et de nanoparticules de SiC a été obtenu. Un traitement post infiltration sous azote, peut être réalisé pour améliorer l’homogénéité du matériau et la stabilité de la microstructure pour des applications sous faibles pressions partielles d’oxygène.
Des gradients de composition ont été détectés à l’interface entre les grains de HfB2 et la « matrice » de SiC. Ils sont associés à la formation d’un eutectique dans le système Hf-Si-B-C, rendu possible par une augmentation de température liée au caractère exothermique de la réaction entre le silicium et le carbone.
Le procédé d’élaboration défini à l’issue de ce travail permet d’obtenir un revêtement homogène et dense de manière reproductible. Une bonne adhésion sur tous les types de substrats testés est obtenue. Cependant, la composition minérale est difficile à évaluer avec précision compte tenu de la complexité des phases formées.
REFERENCES BIBLIOGRAPHIQUES
- AKSAY I. A., HOGE C. E. and PASK J. A., Wetting under chemical equilibrium and nonequilibrium conditions., J. Phys. Chem., 78, 12, 1973, 1178-1183.
- BARTULI C., VALENTE T. and TULUI M., Plasma spray deposition and high temprerature characterization of ZrB2-SiC protective coatings., Surf. Coat. Tech., 155, 2002, 260-273.
- BILL J. and HEIMANN D. Polymer-derived ceramic coatings on C/C-SiC composites. J. Eur. Ceram. Soc., 16, 1996, 1115-1120.
- BROCKMEYER J. W. and WILLIAMS B. E. Characterization and testing of layered HfC/SiC protective coatings. Hard coatings based on borides, carbides & nitrides, 1998, 259-269.
- BRUKL C. E. Part II. Ternary Systems. Volume X. The Zr-Si-C, Hf-Si-C, Zr-Si-B, and Hf-Si-B systems. Ternary phase equlibria in transition metal-boron-carbon-silicon systems, Tech REp. AFML-TR-65-2, 1966.
- CARMAN P. C. Fluid flow through granular beds. Trans. Inst. Chem. Eng., 15, 1937, 168-88.
- CHOE C. R. and HEE LEE K. Effect of processing parameters on the mechnanical properties of carbonized resin. Carbon, 30, 2, 1992, 247-249.
- CLOUGHERTY E. and ANTHONY F. Research and development of refractory oxidation-resistant diborides. Part II Application evaluations and design considerations. Technical report AFML-TR-68-190, 7, 1970
- CLOUGHERTY E., HILL R., RHODES W. and PETERS E. Research and development of refractory oxidation-resistant diborides. Party II Processing and characterisation. Technical report AFML-TR-68-100, 2, 1970
- CLOUGHERTY E., KALISH D. and PETERS E. Research and development of refractory oxidation resistant diborides. Technical report AFML-TR-68-190, 1968
- CLOUGHERTY E., KENNETH E. and RONALD P. Research and development of refractory oxidation-resistant diborides. Part II Thermal, physical, electrical and optical properties. Technical report AFML-TR-68-100, 5, 1969
- DEMIN A., KOSTIKOV V., KRAVETKII G., KUZNETZOV A., RODIONOVA V. and SCHESTAKOVA, 1997, Anti-oxydation protection of carbon-based materials, US5660880, patent
- DULLIEN F. A., EL-SAYED M. S. and BATRA V. K. Rate of capillarity rise in porous media with nonuniform pores. J. of Coll. and Interf. Sci., 60, 3, 1976, 497-506.
- EINSET E. Capillarity infiltration rates into porous media with application to silcomp processing. J. Am. Ceram. Soc, 79, 2, 1996, 333-38.
- EINSET E. O. Anlysis of recative melt infiltration in the processing of ceramic composites. Chem. Eng. Sci., 53, 5, 1998, 1027-1039.
- FAVRE A., BIRKEL T. and FUZELLIER H. Reaction between liquid Al (or Si) and composite C/C materials, Munich. 2001
- FENTER J. R. Refractory diborides as engineering materials.
- FITZER E. and GADOW R. Investigation of the reactivity of different carbons with liquid silicon. Int. Symp. on Ceram. Comp. for Engine. 1983
- GARDZIELLA A., SUREN J. and BELSUE M. Carbon from phenolic resins : Carbon yield and volatile components - Recent studies. Iterceram, 41, 7/8, 1992, 461-467.
- GERN F. Interaction between capillarity flow and macroscopic silicon concentration in liquid silconized carbon/carbon. High temperature ceramic matrix composites II, 149-154.
- GERN F. H. Liquid silicon infiltration : description of infiltration dynamics and silicon carbide formation. Composites Part A, 28A, 1997, 355-364.
- GLASS S. J. and GREEN D. J. Permeability and infiltrattion of partially sintered ceramics. J. Am. Ceram. Soc., 82, 10, 1999, 2745-52.
- GRENIER-LOUSTALOT M.-F., LARROQUE S., GRANDE D. and GRENIER P. Phenolic resins : 2. Influence of catalyst type on reaction mechanisms and kinectics. Polymer, 37, 8, 1996, 1363-1369.
- HASE T. and SUZUKI H. Rise in temperature of SiC pellet involving reaction sintering. J. Nucl. Mat., 59, 1976, 42-48.
- HILLIG W. B. Making ceramic composites by melt infiltration. Am. Ceram. Soc. Bull., 73, 4, 1994, 56-63.
- HILLIG W. B. Melt infiltration approach to ceramic matrix composites. J. Am. Ceram. Soc., 71, 2, 1988, C-96 C-99.
- HOLCOMBE C. E. J. . Slip casting of zirconium diboride, Union Carbides nuclear division. 1972
- HOZER L., LEE J. R. and CHIANG Y. M. Reaction-infiltrated, net shaped SiC composites. Mat. Sci. and Eng., A195, 1995, 131-143.
- KAUFMAN L. . Borides composites - a new generation of nose cap and leading edge materials for reusable lifting re-entry systems. AIAA Advanced Space Transportation Meeting, Cocoa Beach, Florida. 1970
- KIEFFER and BENESOWSKY Powder Metalurgy, (1-2), 1958, 145.
- LACKEY W. J., SMITH A. W. and TWAIT D. Chemical vapor deposition of oxidation resistant HfB2+SiC composite coatings. Ceram. Eng. Proc., 9, 9-10, 1988, 1223-1232.
- LACKEY W., SMITH A., DILLARD D. M. and TWAIT D. Codeposition of dispersed phase ceramic composites. Proc-electrochem Soc., 87-8, 1987, 1008-27.
- LAUSEVIC Z. and MARINKOVIC S. Mechanical properties and chemistry of carbonization of phenol formaldehyde resin. Carbon, 24, 5, 1986, 575-580.
- LUTHRA K. L., SINGH R. N. and BRUN M. K. Toughened silcomp composites- Process and preliminary properties. Am. Ceram. Soc. Bull., 72, 7, 1993, 79-85.
- Mc HALE, A. E. Borides, Carbides and Nitrides. Phase equlibria diagrams. T. A. C. Society., 1994, 10
- MONTEVERDE F. and BELLOSI A. Effect of the addition of silicon nitride on sintering behaviour and microstructure of zirconium diboride. Scr. Metal. Mat., 46, 2002, 223-228.
- MONTEVERDE F., BELLOSI A. and GUICCIARDI S. Processing and properties of zirconium diboride-based composites. J. Eur. Ceram. Soc., 22, 2002, 279-288.
- NECHANICKY M. A., CHEW K. W., SELLINGER A. and LAINE R. M. αSiC/βSiC particulate composite via polymer infiltration and pyrolysis (PIP) processing using polymethylsilane. J. Eur. Ceram. Soc., 20, 2000, 441-451.
- PAMPUCH R., WALASEK E. and BIALOSKORSKI J. Reaction mechanism carbon-liquid silicon systems at elevated temperatures. Ceram. Int., 12, 1986, 99-106.
- RAJESH G. and BHAGAT R. B. Infiltration of liquid metals in porous compacts: modeling of permeabilities during reactive melt infiltration. Transp. in Porous Media, 36, 1999, 43-68.
- REED J. S. Liquid permeability of packed particles : Why perpetuate the Carmen-Kozeny model? J. Am. Ceram. Soc., 76, 2, 1993, 547-48.
- ROMAN-MARTINEZ M. C., CAZORLA-AMOROS D. and LINEARES-SOLANO A. Structural study of phenolformaldehyde char. Carbon, 34, 6, 1996, 719-727.
- SANGSUWAN P., TEWARI S. N., GATICA J. E., SINGH M. and DICKERSON R. Reactive infiltration of silicon melt through microporous amorphous carbon preforms. Metal. Mat. Trans., 30B, 1999, 933-944.
- SINGH M. and BEHRENDT D. R., Reactive melt infiltration of silicon-molybdenum alloys into microporous carbon preforms., Mat. Sci. and Eng., A194, 1995, 193-200.
- SMID, I. and P. ROGL unpublished work. 1987
- TOY C. and SCOTT W. D., Ceramic-metal composite produced by melt infiltration., J. Am. Ceram. Soc., 73, 1, 1990, 97-101.
- TRUMBLE K. P., Spontaneous infiltration of non-cylindrical porosity : close-packed spheres., Acta Mat., 46, 7, 1998, 2363-2367.
- ZHANG G.-J., DENG Z.-Y., KONDO N., YANG J.-F. and OHJI T., Reactive hot pressing of ZrB2-SiC composites., J. Am. Ceram. Soc., 83, 9, 2000, 2330-32