Introduction générale
Ce travail de thèse a porté sur l'implication de l'herpèsvirus humain de type 6 (HHV-6) dans le développement de désordres lymphoprolifératifs et notamment dans celui du lymphome de Hodgkin (LH). L'HHV6 est un virus appartenant à la famille des Herpesviridae et à la sous-famille des b-herpesvirinae. C'est un virus à ADN bicaténaire linéaire présentant une structure icosaédrique. Il est ubiquitaire au sein de la population mondiale. La primo-infection, le plus souvent asymptomatique, peut être caractérisée par une pathologie fébrile du nourrisson : l'exanthème subit dont l'agent étiologique est l'HHV6B. L'HHV6A n'a encore été relié à aucune pathologie. Après la primo-infection, le virus reste dans l'organisme sous une forme de latence fréquemment entrecoupée de périodes de réactivation. Ces réactivations sont, elles aussi, le plus souvent asymptomatiques chez les personnes immunocompétentes mais peuvent être très sévères chez les patients immunodéprimés (transplantés, SIDA). Ce virus pourrait également avoir un lien avec d'autres pathologies dont les étiologies sont encore incertaines comme les désordres lymphoprolifératifs.
Au cours de ce travail, nous nous sommes intéressés à l'association entre l'HHV6 et les lymphomes humains. Nous avons travaillé sur différentes populations de patients atteints de lymphome de Hodgkin, de lymphome B et de lymphome T et NK. Ces pathologies sont des proliférations malignes du tissu lymphoïde, plus ou moins fréquentes suivant la catégorie. Le début de cette étude a porté sur l'ensemble de ces trois catégories, puis a été recentrée sur le lymphome de Hodgkin.
Plusieurs données précédemment publiées par différentes équipes pourraient laisser penser à une implication du virus HHV-6 dans ces désordres lymphoprolifératifs :
- Taux géométriques des anticorps anti-HHV-6 plus élevés chez les patients atteints de lymphome que chez des sujets sains.
- Présence de séquences d'ADN viral dans les adénopathies de ces patients.
- De plus, certaines équipes ont travaillé sur le pouvoir oncogène du virus HHV-6A.
- Caractérisation du pouvoir transformant in vitro de l'HHV6-A.
- Détermination du pouvoir oncogène du fragment d'ADN viral Sal-IL et plus précisément de l'oncogène viral DR7 de l'HHV6-A au sein de ce fragment.
L'objectif de ce travail est d'établir un lien entre l'HHV-6 et les désordres lymphoprolifératifs et plus particulièrement le lymphome de Hodgkin. Pour ce faire, nous avons globalement diviser le travail en cinq grandes catégories.
- Nous avons recherché des séquences d'ADN viral dans les adénopathies de patients atteints de lymphome de Hodgkin, de lymphome B et de lymphome T. Nous avons typé ce virus et déterminé sa charge virale.
- Nous avons également recherché la présence du virus d'Epstein-Barr dans les adénopathies des différents patients atteints de lymphomes.
- Nous avons séquencé l'oncogène viral DR7 présent dans les adénopathies des patients atteints de lymphomes afin de mettre en évidence d'éventuelles mutations pouvant être spécifiques d'une ou de plusieurs de ces pathologies.
- Nous avons travaillé sur l'altération de la p53 dans les phénomènes de cancérisation. Nous avons séquencé les exons 5 à 8 du gène de la p53 dans les adénopathies des patients atteints de lymphome de Hodgkin. Nous avons, ensuite, déterminé la capacité de la protéine virale DR7 à se lier à la p53 humaine.
- Finalement, nous avons voulu savoir si l'HHV-6, et plus particulièrement l'oncoprotéine virale DR7 et la protéine transactivatrice virale U3, étaient impliqués dans l'activation constitutive du facteur de transcription NFκB, décrite au niveau du lymphome de Hodgkin.
Environnement bibliographique - Introduction
1. Herpèsvirus humain de type 6 (HHV-6)
1.1. Description générale :
L'herpèsvirus humain de type 6 (HHV-6) est un virus découvert en 1986 par Salahuddin (1). Il fut isolé fortuitement à partir de cellules mononucléées de sang périphérique (PBMC) chez des patients atteints du SIDA ou de désordres lymphoprolifératifs. Précédemment appelé HBLV pour Human B Lymphotropic Virus, il fut rapidement rebaptisé HHV-6 lorsque son appartenance à la famille des Herpesviridae et que son tropisme pour les cellules T furent mis en évidence. Ce virus appartient à la sous-famille des b-herpesvirinae comme l'herpèsvirus humain de type 7 (HHV-7) et le cytomégalovirus humain (CMV) (Tableau 1). Il existe deux types d'HHV-6, A et B, présentant des différences reposant sur des critères de croissance virale, d'épidémiologie, de séquences génomiques et de propriétés antigéniques (2). L'HHV-6A, l'HHV-6B et l'HHV-7 composent le genre des roséolovirus de la sous-famille des b-herpesvirinae.
1.2. Souches virales de référence :
Il existe plusieurs souches virales de référence ; pour le type A, les souches U1102 et GS ; pour le type B, les souches Z29 et HST. La souche prototype U1102 de laboratoire a été isolée d'un patient Ougandais atteint du SIDA (3). Les séquences génomiques de cette souche ont été décrites par Gompels et al. en 1995 (4) (GENBANK n° NC 001664). Le génome de la souche GS de l'HHV-6A, provenant de l'isolat initial de Salahuddin, n'a pas encore été séquencé.
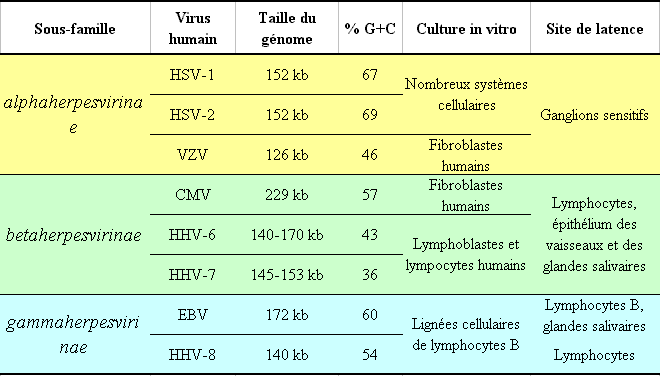
HSV 1: herpès simplex virus 1, HSV 2: herpès simplex virus 2, VZV : virus de la varicelle et du zona, HHV-7 : herpèsvirus humain de type 7, EBV : virus d'Epstein Barr, HHV-8 : herpèsvirus humain de type 8.
La souche HHV-6B HST a été isolée à partir d'un patient atteint d'un exanthème subit (5) et la souche Z29 d'un patient atteint du SIDA au Zaïre (6). Le génome de ces deux souches a été entièrement séquencé par deux équipes différentes en 1999 (7, 8) ; leurs séquences sont déposées dans les banques de gènes (GENBANK n° AB021506 et n° NC 000898).
1.3. Tropisme cellulaire :
In vitro, l'HHV-6A et l'HHV-6B se multiplient plus efficacement dans les lymphocytes T primaires activés. Ces virus poussent relativement mal sur lignées cellulaires et l'HHV-6B cultive encore moins bien que l'HHV-6A, mais plusieurs isolats ont été tout de même adaptés à la culture sur lignée cellulaire T transformée. L'HHV-6A souche GS est le plus souvent cultivée sur la lignée humaine lymphoblastoïde T HSB2 alors que la souche U1102 l'est sur les cellules humaines T lymphoblastoïdes J JHAN. La souche Z29 de l'HHV-6B a également été adaptée à la culture sur lignée humaine de lymphocytes T Molt 3 (provenant d'une leucémie aiguë lymphoblastique T) ou les cellules humaines lymphoblastoïdes T MT4 pour l'HHV-6B souche HST.
Bien que les cellules T soient les plus utilisées pour la propagation virale, des lignées cellulaires comme les mégacaryocytes, les glioblastomes, les cellules neurales, épithéliales et les fibroblastes sont également sensibles à l'infection par l'HHV-6 mais la réplication virale est souvent plus faible (9-12).
In vivo, les types de cellules hôtes sont beaucoup plus nombreux. L'HHV-6 possède un tropisme marqué pour les cellules T CD4+ (13, 14), mais le virus peut aussi se répliquer dans les cellules T CD8+, les cellules B, les cellules NK (natural killer) (15), les monocytes/macrophages (16), les cellules endothéliales tubulaires rénales (17), les glandes salivaires (18). Des protéines virales ont été également détectées dans des neurones et des oligodendrocytes (19, 20).
L'infection par l'HHV-6 induit un effet cytopathogène des cellules infectées qui leur confère un aspect ballonnisé.
1.4. Structure :
Les particules virales ont une dimension comprise entre 160 et 200 nm et présentent une morphologie typique des herpèsvirus (Figure 1), constituée de 4 éléments structuraux principaux :
- une nucléocapside,
- un core,
- un tégument,
- une membrane.
La nucléocapside à symétrie icosaédrique de 90 à 110 nm de diamètre contenant 162 capsomères, enferme un ADN bicaténaire linéaire, à l'intérieur d'un core dense aux électrons. Cette capside est elle-même entourée par une bicouche lipidique dérivant de membranes cellulaires de la cellule hôte et au sein de laquelle sont ancrées les glycoprotéines virales. Une matrice protéique peu structurée appelée tégument occupe l'espace entre la nucléocapside et l'enveloppe externe (Figure 1) (21, 22).
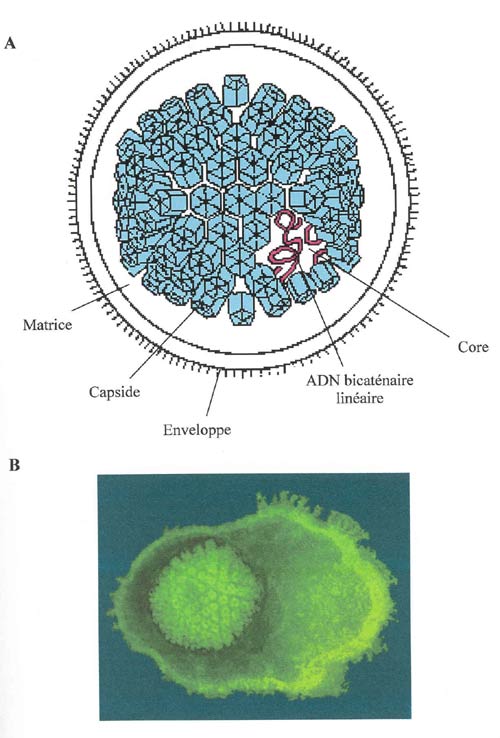
(A) Schéma de la structure classique des herpèsvirus (B) Photo en microscopie électronique d'une particule de l'herpèsvirus simplex de type 1 (HSV1).
Site Internet : http://web.uct.ac.za/depts/mmi/stannard/herpes.html.
1.5. Organisation génomique :
Le génome de l'HHV-6 (Figure 2) est composé d'une molécule d'ADN bicaténaire linéaire d'environ 160 kb pour le type B (7, 8) et 140 kb pour le type A (4). L'architecture génomique de l'HHV-6 est également trouvée chez l'HHV-7 mais aussi chez le virus du poisson chat (Catfish virus).
Pour les deux types (A et B), le génome viral comporte les 7 blocs de gènes conservés au sein des Herpesviridae (I à VII), un bloc caractéristique des _-herpesvirinae (U2 à U19), une séquence interne répétée (IR), et à chaque extrémité, une séquence appelée DR pour Direct Repeat, constituée de motifs directement répétés (Figure 2). Les types A (U1102) et B (Z29) présentent 119 cadres ouverts de lecture (ORF : Open Reading Frame) (4, 7), alors que la souche B HST présente 115 ORF (8). La longueur des DR peut également varier de 8 à 13 kb en fonction du nombre de passages du virus en culture in vitro. Le pourcentage de GC ne semble pas constant tout au long du génome avec un pourcentage moyen plus élevé au niveau des DR qu'au niveau de la séquence unique. La très grosse majorité de l'ADN de ce virus est codant. Les cadres de lecture situés dans les DR portent le préfixe DR et ceux situés dans la région unique sont nommés U1-100 partant de la gauche jusqu'à la droite du génome. Les gènes de l'HHV-6A et de l'HHV-6B appartenant aux 7 blocs présentent jusqu'à 94% d'identité (23). La comparaison des génomes de l'HHV-6A et de l'HHV-6B confirme qu'ils sont colinéaires avec environ 90% d'identité. Les régions présentant des variations significatives sont situées dans les DR ainsi que dans une région de 24 kb localisée à la droite de U86.
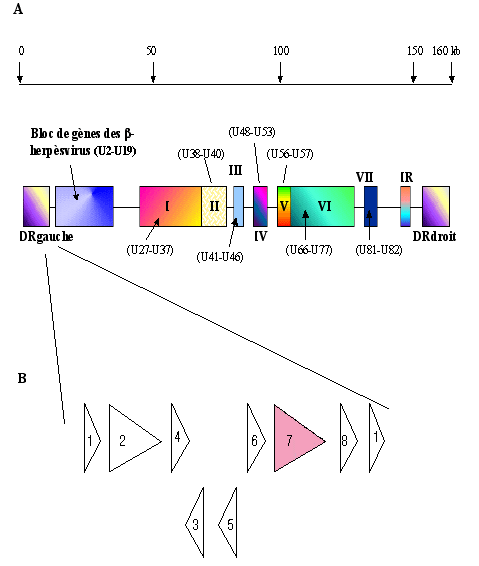
(A) Génome de l'HHV-6B souche Z29 (Dominguez et al., 1999). DR : séquence directe répétée, IR : séquence inversée répétée. (B) Composition génétique des DR (DR1-DR8) (Gompels et al., 1995 ; Dominguez et al., 1999).
Chaque DR présente 8 cadres ouverts de lecture différents entre eux mais identiques entre le DRgauche et le DRdroit (Thompson et al., 1994 ; Kashanchi et al., 1997).
Le rôle des DR est encore incertain. Ils seraient impliqués dans la circularisation du virus lors de l'infection productive. Il existe dans des DR des séquences homologues de signal de clivage/empaquetage : pac-1 et pac-2 (24). Les DR interviendraient également dans la réplication virale (25). Des séquences télomériques humaines (GGGTTA)n ont été détectées au niveau des DR (24). Ces séquences télomériques sont également présentes au niveau du génome intégratif de l'herpèsvirus aviaire provoquant la maladie néoplasique de Marek chez les poulets (26). Ces différentes données ont permis à certains auteurs d'envisager une implication de ces séquences nucléotidiques dans une éventuelle intégration du génome viral dans des chromosomes des cellules mononucléées. Mais cette hypothèse reste controversée. En effet, quelques études seulement ont pu mettre en évidence une intégration de ce virus. L'équipe de Luppi et al. ont pu mettre en évidence 3 cas d'intégration du génome viral. Deux patients atteints de désordres lymphoprolifératifs et un patient atteint de sclérose en plaque présentaient une intégration complète du génome viral (27). Des études plus récentes réalisées par cette même équipe ont démontré une intégration virale ciblée. En effet, ces 3 patients présentaient un site d'intégration sur l'extrémité télomérique du brin court du chromosome 17 (17p13.3) (28). Ce site d'intégration est relativement proche de deux oncogènes cellulaires : CRK et ABR pouvant éventuellement entraîner une dérégulation de ces gènes (29). L'équipe de Daibata a également pu mettre en évidence une intégration du génome viral dans une lignée cellulaire établie à partir des lymphocytes d'un patient atteint d'un lymphome de Burkitt (EBV négatif) au niveau du bras long du chromosome 22 (22q13) (30) ainsi qu'une intégration du génome viral au niveau du locus 1q44 dans les cellules du sang périphérique chez un sujet sain (31). Ils ont également mis en évidence une transmission héréditaire de ces 2 intégrations virales chez la fille de ce couple. Ces quelques cas ne sont pas suffisants pour envisager une intégration fréquente du génome de l'HHV-6. Chez ces patients étudiés, on observe également des sites d'intégrations différents. Il pourrait s'agir simplement de cas isolés ne reflétant pas le comportement général du virus.
1.6. Cycle viral :
La physiopathologie des infections à herpèsvirus consiste en une phase lytique avec une production de nouvelles particules virales (primo-infection ou réactivation) suivie d'une phase de latence caractérisée par l'absence de production de particules virales. Le cycle d'infection lytique de l'HHV-6 est décrit classiquement comme la succession de différentes étapes (Figure 3). L'attachement de la particule infectieuse à la membrane plasmique s'effectue à l'aide du récepteur cellulaire cofacteur de la membrane CD46 (32). Le CD46 appartient à la famille des régulateurs de la fixation du complément. Santoro et al. ont démontré que la protéine CD46 était essentielle mais pas suffisante pour l'entrée du virus dans la cellule. La protéine CD46 étant présente à la surface de toutes les cellules nucléées (33, 34), l'auteur a envisagé l'intervention d'un facteur cellulaire supplémentaire comme un co-récepteur expliquant que le virus n'infectait pas toutes les cellules porteuses du CD46. Mori et son équipe ont montré que le complexe viral glycoprotéine H (gH)- glycoprotéine L (gL) était un ligand privilégié pour le récepteur CD46 pour l'entrée du virus dans la cellule. Ils ont également démontré que le produit du gène d'HHV-6A U100 appelé glycoprotéine Q (gQ) se fixait au complexe gH-gL formant un trimère ligand de CD46 (35, 36). Dans une étude très récente, ils ont aussi démontré que le complexe gH-gL-gQ de l'HHV-6B n'avait pas la capacité de se lier à CD46, pouvant expliquer la différence de tropisme cellulaire entre ces deux virus (Communication orale, abstract 4.04, 28th international herpesvirus workshop, Madison, 2003).
L'étape de pénétration de la capside au sein du cytoplasme se fait par fusion de l'enveloppe virale avec la membrane plasmique. La capside est ensuite dirigée par le réseau microtubulaire jusqu'à un pore nucléaire au travers duquel l'ADN est injecté dans le noyau où il est transcrit et répliqué (Figure 3). Les différentes protéines nécessaires aux virus sont alors synthétisées pendant que l'ADN viral est répliqué. La réplication de l'ADN viral génère de façon efficace un nombre élevé de copies d'ADN viral. L'assemblage des protéines de la capside a ensuite lieu dans le noyau autour de génomes nouvellement formés et clivés produisant des particules mûres. Ces vacuoles fusionnent avec la membrane cellulaire, entraînant l'enveloppement des particules, et le bourgeonnement à la surface cellulaire afin de libérer les particules virales provoquant la lyse de la cellule hôte (Figure 3).
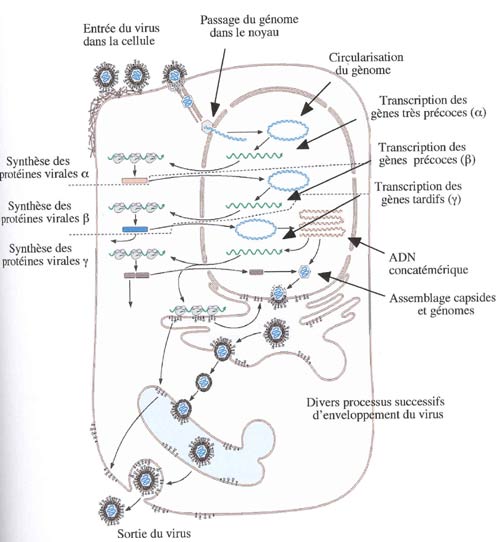
Figure trouvée sur le site Internet http://www.edumed.ch/apprentissage/virolab/bibliothèque et modifiée.La circularisation du génome semble être une étape nécessaire pour la réplication de l'ADN des herpèsvirus. Elle pourrait s'effectuer par liaison directe des terminaisons génomiques gauche et droite (37-39). Mais la recombinaison homologue entre les séquences terminales répétées pourrait également être à la base de la circularisation du génome (40). La synthèse d'ADN commence au niveau de l'origine de réplication oriLyt, localisée entre l'extrémité 5' du gène U41 et l'extrémité 3' du gène U42, entre les positions 67617 et 67993 pour l'HHV-6A U1102 (4, 41). Le segment oriLyt minimal mesure environ 300 pb, mais le segment pleinement fonctionnel mesure environ 800 pb (41). Il conduirait à la formation de structures appelées concatémères consistant en la liaison covalente d'unités génomiques dans un réarrangement tête-à-queue (42). Le rôle précis de cette séquence est encore mal connu. Elle comprend au centre un élément majeur composé de 2 séquences palindromiques OBP-1 et OBP-2 qui sont reconnues par la protéine OBP (Origin Binding Protein). Par analogie avec l'herpès simplex virus humain (HSV1), l'initiation de la réplication devrait faire intervenir les protéines OBP fixées aux 2 sites OBP (43).
Durant l'infection lytique, il existe une expression cinétique de 3 classes de gènes. Les premiers ARN transcrits (apparaissant environ 3 heures après l'infection) correspondent aux gènes très précoces ( _ou IE pour Immediate Early) sont les gènes U42, U89-90, U94, et U73. Beaucoup de gènes IE codent des transactivateurs de gènes viraux plus tardifs. La transcription des gènes IE ne nécessite pas de synthèse de protéines de novo. Les gènes précoces ( _ ou E pour Early) sont ensuite transcrits aboutissant à des produits intervenant dans le métabolisme de réplication de l'ADN. Ce sont les gènes U19, U31, U41 et U53 qui sont détectés 8 heures après le début de l'infection. La transcription des gènes E est dépendante de l'activité des gènes IE. Enfin les gènes tardifs (g ou L pour Late) codent principalement des protéines structurales (les glycoprotéines d'enveloppe, les protéines du tégument, les protéines majeures de la capside) et des enzymes responsables du métabolisme (44). La transcription des gènes L est dépendante de la réplication de l'ADN viral.
La détection entre un état latent ou un état réplicatif du virus peut être déterminée par la présence de transcrits très précoces, précoces ou tardifs. Il existe un certain nombre de techniques de RT-PCR sur des transcrits très précoces (U94 et U90) et sur des transcrits plus tardifs (U31 : protéine large du tégument et U39 : glycoprotéine B) (45). Le détail de la technique est étudié dans le chapitre diagnostic.
L'HHV-6 possède un certain nombre de transactivateurs viraux dont les rôles sont plus ou moins bien connus. Ils interviendraient dans la régulation de la transcription des gènes viraux mais aussi cellulaires. Les protéines transactivatrices virales sont le plus souvent codées par des gènes IE ou E. L'HHV-6 présente parmi ces transactivateurs 11 membres appartenant à la famille des transactivateurs US22 (homologues du CMV), DR1, DR2, DR6, DR7, U2, U3, U7, U8, U16, U25 et U95. Cette famille de gènes US22 est présente chez les b-herpèsvirus mais pas chez les a et g -herpèsvirus.
Le gène viral U3 appartenant à la famille des gènes US22 (homologues du CMV), code une protéine transactivatrice pouvant être traduite à partir de deux transcrits, un de 2 kb et l'autre de 3,5 kb. Ces deux ARNm présentent la totalité de la séquence codante sans aucun épissage. Par immunofluorescence, les antigènes U3 sont détectés dans les 24 h suivant l'infection des cellules U373 par l'HHV-6A. Par des techniques de co-transfections avec expression de gènes rapporteurs de luciférase, Mori et al. ont démontré que la protéine U3 avait la capacité d'activer les LTR (Long Terminale Repeat) du VIH-1 dans les cellules CV1 (46).
Le gène DR7 appartient également à la famille des transactivateurs US22. Les ARNm de DR7 sont détectés dans une culture virale de l'HHV6-A souche U1102 à partir de 18h jusqu'à 72h après infection, avec une transcription des ARNm et une traduction protéique simultanées (47). La protéine DR7 de l'HHV-6A souche U1102 a la capacité de transactiver in vitro les LTR du VIH aboutissant à une augmentation de la production virale (48). De plus, les cellules exprimant la protéine DR7 sont tumorigènes quand elles sont injectées dans des souris nude alors que des cellules produisant une protéine tronquée ne le sont pas (47).
La protéine IE-2 de l'HHV-6A souche GS est également décrite comme transactivatrice. L'infection virale induit la synthèse d'un ARNm IE-2 de 5,5 kb, 2 à 4 h après l'infection virale avec accumulation jusqu'à 96h. La protéine de 200 kDa réagit comme un activateur transcriptionnel induisant la transactivation des LTR du VIH mais également celle de promoteurs contenant les éléments de réponse CRE (élément de réponse à CREB), NFAT, NFkB ou une simple boîte TATA, jouant probablement un rôle important dans l'initiation de l'expression de gènes de l'HHV-6 (49).
Le gène U42 est également décrit comme transactivateur. Il code un homologue du gène IE d'HSV a27 et du gène E du CMV UL69. Le gène U42 n'appartient pas à la famille US22. Il s'agirait d'un gène appartenant à la catégorie E (gènes précoces) mais il ne semble pas transcrit en l'absence de synthèse de protéines de novo (50).
L'encapsidation se déroule, comme pour les autres herpèsvirus, dans le noyau de la cellule infectée, où l'assemblage de la capside et la réplication de l'ADN viral ont également lieu. L'encapsidation nécessite la présence de l'ADN concatémérique, des capsides synthétisées et de certaines protéines virales. Des protéines interviennent dans le clivage de l'ADN concatémérique au niveau de sites spécifiques de façon à encapsider un génome viral complet. Il existe des sites spécifiques au niveau du génome viral, pac1 et pac2, nécessaires aux clivages et à l'encapsidation de l'ADN viral (51). Des capsides contenant l'ADN viral apparaissent environ 3 jours après le début de l'infection (52). Les molécules d'ADN nouvellement synthétisées vont donc se lier aux protéines de capside pour former la nucléocapside. Cet assemblage a lieu avant l'acquisition du tégument dans les compartiments nucléaires aboutissant à la formation des tégusomes. Après fusion du tégusome avec la membrane nucléaire, les nucléocapsides sont relarguées dans le cytoplasme.
La libération des particules virales a lieu après acquisition de l'enveloppe. Plusieurs voies existent selon les herpèsvirus. L'HSV1 acquiert son enveloppe en passant du noyau vers le cytoplasme. Le virus de la varicelle et du zona (VZV) et les roséolovirus utiliseraient des processus successifs d'enveloppement, de désenveloppement et de ré-enveloppement en fonction des passages des particules virales d'un compartiment à l'autre. Les particules virales sont enfin relarguées par exocytose ou lyse cellulaire. (53). Dans une culture, des virions matures apparaissent environ 5 jours après le début de l'infection (52).
1.7. Effet du virus sur la cellule hôte :
L'infection provoquée par les roséolovirus induit de profonds changements sur les cellules hôtes comme une margination de la chromatine (54), un arrêt de la synthèse d'ADN cellulaire pour l'HHV-6B dans les 65h après l'infection (55), alors que l'HHV-6A n'entraînerait pas d'arrêt de la synthèse protéique cellulaire mais plutôt une stimulation généralisée de la synthèse de protéines (56, 57). Cela aboutit au développement d'effets cytopathogènes classiques de ballonnisation, donnant des cellules infectées géantes et multinucléées formant des syncytia.
1.8. HHV-6 et pathologies :
1.8.1. Epidémiologie et transmission virale :
L'HHV-6 est un virus ubiquitaire : plus de 95% de la population mondiale adulte est séropositive pour ce virus (types A ou B ou les deux). La transmission virale a lieu dans la petite enfance et paraît se faire principalement de la mère à l'enfant ou entre enfants, le plus souvent via la salive (18). Une autre transmission possible de l'HHV-6 est la voie intra-utérine. Des séquences d'ADN viral ont été détectées dans un foetus et dans le sang d'un nouveau-né (58-60). La réactivation de l'HHV-6 semble être un événement courant pendant la grossesse et le transfert du virus au foetus paraît possible bien que probablement très rare. D'autres voies probables de transmission ont été décrites comme lors de transplantations (61).
1.8.2. Infections virales :
1.8.2.1. Primo-infections :
L'infection primaire a lieu le plus souvent entre 6 mois et 2 ans (62). L'HHV-6 infecte les enfants le plus souvent de manière asymptomatique. Néanmoins, dans certains cas, l'HHV-6B provoque l'exanthème subit qui est une pathologie fébrile du nourrisson (5) se caractérisant par une fièvre de 3 à 5 jours s'accompagnant d'un rash cutané, par contre la primo-infection à HHV-6A n'a encore été reliée à aucune pathologie. Il existe des primo-infections plus tardives chez certains adultes n'ayant pas été contaminés enfants, et la primo-infection chez l'adulte est souvent plus grave que chez l'enfant. Chez ces patients, nous pouvons observer des syndromes mononucléosiques parfois être sévères. De même l'HHV-6 a été associé à de rares cas d'hépatites fulminantes graves aussi bien chez l'adulte que chez l'enfant (63, 64).
1.8.2.2. Persistance virale :
Après la primo-infection, le virus reste dans l'organisme dans un état de latence régulièrement entrecoupé de périodes de réactivation. L'infection persistante peut être classifiée en infection latente et en infection chronique. Pendant l'infection latente, il n'y a pas de production de particules virales, l'expression des gènes viraux se limite à celle des gènes requis pour le maintien de la latence comme le gène U94 (65). Alors que pendant l'infection chronique, des particules virales sont produites pouvant aboutir à une transmission virale.
Après la primo-infection, le virus reste latent dans l'organisme probablement dans les monocytes/macrophages (16) mais aussi dans les cellules progénitrices de la moelle osseuse (66). L'HHV-6 paraît également persister dans les cellules épithéliales des glandes salivaires (où il se réplique lors des périodes de réactivation) (67) dans les neurones et dans les oligodendrocytes du système nerveux. Cette latence est entrecoupée régulièrement d'épisodes de réactivation, le plus fréquemment sans manifestations cliniques. Par contre, l'HHV-6 entraîne, chez les patients immunodéprimés, des pneumopathies sévères voire mortelles (68, 69), des rétinites chez les patients atteints du SIDA (70) et des lymphomes. Chez les patients transplantés, la réactivation de l'HHV-6 peut aboutir à des pneumopathies, des encéphalites (71), des fièvres, pouvant compliquer la transplantation voire même entraîner un rejet de la greffe et la mort du patient.
L'HHV-6 a été incriminé parmi les causes du syndrome de fatigue chronique (72, 73). Yalcin et al. ont démontré la présence de séquences du génome viral dans les PBMC de 7/13 (soit 53%) des patients atteints du syndrome de fatigue chronique (73).
De même, un lien a été suspecté entre l'HHV-6 et maladies auto-immunes neuro-dégénératives comme la sclérose en plaque (19). Challoner et al. ont démontré que le virus de l'HHV-6 était présent dans le cerveau de plus de 70% des cas de patients atteints de sclérose en plaque avec une grande majorité de type B (19). Goodman et al. ont déterminé la présence du virus dans les oligodendrocytes au niveau de toutes les lésions chez 5 patients atteints de sclérose en plaque, prouvant que l'HHV-6 pouvait être impliqué dans le phénomène de démyélinisation rencontrée lors de cette pathologie (74).
Le virus pourrait également jouer un rôle étiologique dans la survenue de certains désordres lymphoprolifératifs. L'HHV-6 est souvent trouvé dans les adénopathies de patients atteints de lymphome de Hodgkin, mais en ce qui concerne les lymphomes non Hodgkiniens (LNH), la présence du virus est plus variable, vue la grande diversité des pathologies regroupées dans cette catégorie. Les différentes études effectuées sur la présence de l'HHV-6 dans les lymphomes de Hodgkin seront détaillées plus loin et celles effectuées sur les lymphomes non hodgkiniens sont répertoriés dans le tableau 2.
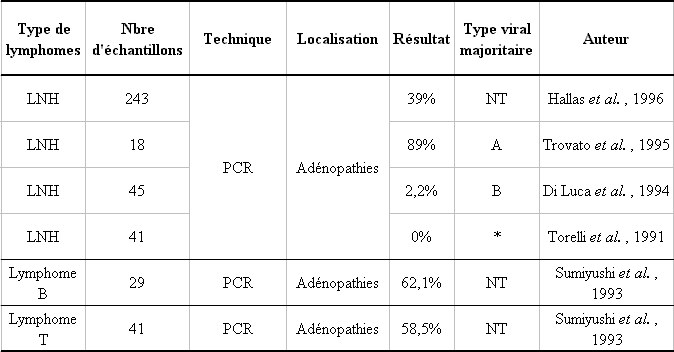
LNH : lymphome non hodgkinien, NT : non testé.
Il est difficile de tirer des conclusions au vu de l'ensemble des résultats du tableau 2, quant à la présence systématique du virus dans les adénopathies des patients atteints de lymphome non hodgkinien (LNH). Les séquences d'ADN du virus ont été détectées à partir de 0% jusqu'à 89% des prélèvements provenant de patients atteints de LNH, avec des pourcentages de détection soit très faibles (0 et 2,2%) ou relativement élevés (62,1% et 89%) (75-79). Il ne semble pas exister de différences de détection du virus de l'HHV-6 entre les lymphomes B et les T (62,1% et 58,5% respectivement) (76).
L'étude réalisée par Ohyashiki et al. a permis de déterminer la charge virale de l'HHV-6 dans les biopsies de 19 patients atteints de désordres lymphoprolifératifs. Ils ont trouvé que le virus était présent dans quasiment tous les lymphomes B testés mais avec une charge virale inférieure à 5 copies virales par mg d'ADN, par contre elle est beaucoup plus élevée chez deux patients, l'un atteint d'une lymphadénopathies immunoblastique et l'autre atteint d'un lymphome T (3705 et 810 copies virales pour 1 κg d'ADN respectivement). D'après cette étude, les auteurs ont conclu que les charges virales élevées étaient une exception pour les cas de LNH (80).
1.9. Diagnostic :
Le diagnostic de l'HHV-6 peut s'effectuer à l'aide de différentes techniques :
La sérologie présente un intérêt diagnostic modeste du fait de la très large diffusion de ce virus dans la population générale. La présence des IgM est fréquemment interprétée comme indicatrice d'une primo-infection ou d'une réactivation virale. Or pour l'HHV-6, la détection d'IgM ne peut être systématiquement corrélée à une réactivation virale. En effet, Suga et al. ont détecté l'apparition d'IgM chez des enfants avec un exanthème subit entre le 5e et le 7e jour de la maladie, avec un titre maximum entre la 2e et la 3e semaine et un déclin jusqu'à l'obtention de taux indétectables après deux mois. Ils ont aussi montré que chez des patients transplantés présentant une réactivation de l'HHV-6, les taux d'IgM détectées étaient beaucoup plus élevés, persistant pendant 2 à 3 mois et devenant indétectables 5 à 6 mois après la transplantation. Ils ont également cherché la prévalence des IgM anti-HHV-6 chez des sujets sains. Les anticorps ont été détectés chez 5% des enfants entre 4 et 7 mois, chez 40% entre 8 et 11 mois (taux très élevés) et chez 17% entre 4 et 7 ans (taux juste détectables) (81).
Par contre, la recherche des IgG anti HHV-6 est intéressante dans les cas des séroconversions chez les enfants. Ueda et al. ont décrit que 43/53 (81,1%) des enfants présentant un exanthème subit montraient une séroconversion (82).
La culture du virus est une technique assez peu utilisée pour le diagnostic. Au cours de la primo-infection, l'HHV-6 peut être cultivé à partir de lymphocytes d'enfants pendant la phase fébrile de l'exanthème subit (83). Elle s'effectue par mise en contact avec des lymphocytes périphériques sanguins (sang de donneur adulte ou sang de cordon) stimulés par de la phytohémagglutinine pendant 48h. Les cellules sont alors cultivées en présence d'interleukine 2 (IL-2), de polybrène et de sérum anti-interféron. Le suivi de l'infection s'effectue le plus souvent par observation des cultures au microscope dans le but de détecter un effet cytopathique (84). En revanche, certaines souches n'entraînent pas toujours d'effet cytopathique, il devient, par conséquent, nécessaire de détecter soit des antigènes, soit le génome viral (84).
La recherche des antigènes d'HHV-6 s'effectue à l'aide d'anticorps monoclonaux spécifiques. Il existe un certain nombre d'anticorps monoclonaux, notamment dirigés contre la glycoprotéine B, mais la majorité d'entre eux ne reconnaissent spécifiquement que l'un ou l'autre des types A ou B (85). Par exemple, l'anticorps monoclonal 6E2 est spécifique de l'HHV-6 type B mais ne réagit pas avec les prototypes U1102 et GS du type A (86). Cette technique peut être pratiquée pour le typage des résultats obtenus lors de la culture.
La recherche du génome de l'HHV-6 peut être réalisée par hybridation in situ ou par amplification génique (polymerase chain reaction, PCR). La détection du virus dans le sérum ou le plasma permet de diagnostiquer une infection active à HHV-6. De nombreuses techniques de PCR ont été développées afin de détecter le génome viral. Cone et son équipe ont développé une technique de PCR suivie de l'hybridation d'une sonde sur l'amplicon du gène viral U67. Cette méthode de détection est très sensible et hautement spécifique (87). La PCR semble la méthode de détection la plus répandue à l'heure actuelle.
Finalement, la distinction entre un état réplicatif ou latent du virus peut se faire par la recherche d'ARNm viraux tardifs ou par des techniques de PCR quantitative. Secchiero et al. ont développé une méthode de PCR quantitative par compétition pour les virus HHV-6 et HHV-7, de façon à quantifier simultanément les deux génomes viraux (88). De même, Clark et son équipe ont mis au point une PCR quantitative compétitive pour le virus HHV-6 (89). Les techniques de PCR quantitative compétitive n'étaient pas très développées du fait de la complexité de la mise au point. De nouvelles technologies, les PCR quantitatives en temps réel, se sont développées assez récemment et ont permis de révolutionner la PCR quantitative. Gautheret-Dejean et ses collaborateurs ont mis au point une technique de PCR quantitative pour le virus HHV-6 suivant la technologie TaqMan. Cette méthode permet la détection indifféremment les deux types A et B, par l'amplification d'un fragment amplifié de la région U65-U66 du génome d'HHV-6 (90).
Yoshikawa et al. ont récemment développé une technique de transcription inverse suivie d'une PCR dans le but de suivre l'infection par l'HHV-6 et de distinguer un virus réplicatif d'un virus latent. Ce test repose sur la détection des ARN messagers de 4 gènes viraux différents : U31 et U39 (gènes tardifs) et U90 et U94 (gènes très précoces) (45). Le gène U94 est associé à la latence, seul son transcrit étant détecté pendant la phase de latence de ce virus (65). Les auteurs ont démontré que les trois transcrits des gènes U31, U39 et U94 étaient présents dans les PBMC de 90% à 100% des échantillons collectés chez des enfants infectées par l'HHV-6 pendant la phase fébrile de l'exanthème subit. Les transcrits des gènes U31 et U39 n'étaient plus présents dans les échantillons pendant la période de convalescence ne contenant plus de virus infectieux. Le transcrit d'U94 décrit comme associé à la latence est détecté dans 10% des échantillons pendant cette période et le transcrit d'U90 dans 15% des échantillons. Par contre, dans les PBMC de patients présentant une réactivation virale, les transcrits des gènes viraux U31 et U94 n'ont pas été détectés (45).
1.10. Traitements :
1.10.1. Descriptions des drogues antivirales :
Il existe différentes catégories d'antiviraux utilisés contre les herpèsvirus humains avec des modes d'action différents les uns des autres. Les traitements antiviraux utilisés contre les herpèsvirus sont souvent à base d'analogues nucléosidiques. Tous les nucléosides artificiels qui inhibent la multiplication des virus sont des promédicaments. Inactifs, ils doivent généralement être triphosphorylés avant d'exercer leur activité antivirale. Ces phosphorylations sont réalisées par des kinases, le plus souvent, en trois étapes successives (91). La première étape de phosphorylation est cruciale, elle doit être réalisée par des kinases virales. Les deux autres étapes de phosphorylation sont effectuées par des kinases cellulaires (92). Le mécanisme peut être un peu différent suivant les drogues utilisées (Figure 4). Citons quelques exemples :
- la dihydropropoxyméthyl-guanine
(DHPG, ganciclovir, GCV, CymevanTM) est un analogue nucléosidique
de la guanine. Son action antivirale est liée à une inhibition sélective
de la synthèse d'ADN viral, par la forme triphosphate (Figure 4) du produit,
à deux niveaux,
- inhibition compétitive de l'ADN polymérase virale,
- inhibition de l'élongation de l'ADN (84).
- l'hydroxyphosphonylméthoxypropyl-cytosine (HPMPC, cidofovir, CDV, VistideTM) est un analogue nucléosidique de la cytosine. Contrairement aux analogues nucléosidiques comme le GCV, le CDV ne nécessite pas de phosphorylation initiale dépendante des enzymes virales. Les deux phosphorylations dépendent des enzymes cellulaires (Figure 4). Le CDV diphosphate inhibe l'ADN polymérase en s'intégrant dans la chaîne d'ADN lors de l'élongation (84).
- l'acycloguanosine (acyclovir, ACV ou ZoviraxTM) est également un analogue nucléosidique de la guanine. Sa forme active est triphosphorylée, avec une première phosphorylation réalisée par une enzyme virale et les deux autres par des enzymes cellulaires (Figure 4). L'ACV inhibe de façon compétitive l'ADN polymérase et entraîne un blocage de la chaîne d'ADN viral en cours de synthèse en raison de son incapacité à former une liaison phosphodiester 3'5' avec un autre nucléoside triphosphaté (91).
Après leurs phosphorylations, les analogues de structure (GCV, CDV, ACV) entrent en compétition avec leurs homologues naturels, en tant que substrat de l'ADN polymérase virale. Par leur présence, ils bloquent le site actif et/ou s'incorporent dans le brin d'ADN néoformé, créant des modifications de séquences. Ces dernières entraînent l'arrêt de la réplication, ou la transcription d'ARNm anormaux non traduisibles en protéines actives (GCV).
- l'acide phosphonoformique (Foscarnet, PFA, FoscavirTM) est un analogue des pyrophosphates. Il inhibe sélectivement la réplication virale dans les cellules infectées, sans atteindre les cellules normales. Il agit en bloquant directement, et de façon non compétitive, le site du récepteur au pyrophosphate de l'ADN polymérase virale. L'activité du PFA ne nécessite pas de phosphorylation intracellulaire (84).
De nombreuses nouvelles molécules antivirales sont en cours de développement. Il existe déjà sur le marché une prodrogue du GCV qui est le GCV valiné ou Valganciclovir (ValcyteTM). Son action est similaire à celle du GCV. Il existe aussi l'ACV valiné ou valaciclovir (ZélitrexTM). Ces drogues permettraient de mieux cibler les cellules infectées et par conséquent de rendre un traitement plus efficace.
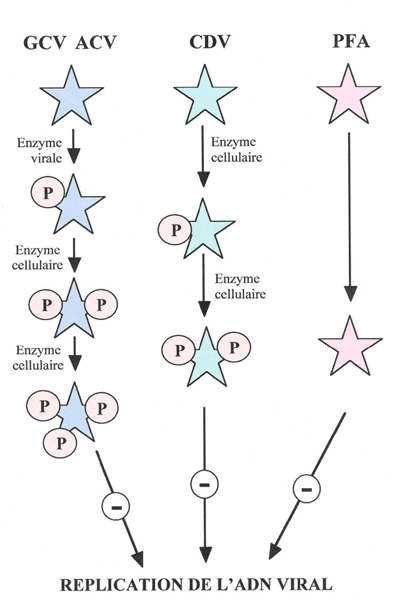
GCV : ganciclovir, ACV : acyclovir, CDV : cidofovir, PFA : foscarnet.
1.10.2. Etudes réalisées sur la sensibilité de l'HHV-6 aux traitements antiviraux :
Le GCV, le PFA ainsi que le CDV sont des inhibiteurs potentiels de la réplication des roséolovirus. Plusieurs études ont été réalisées sur la sensibilité de l'HHV-6 aux différents drogues utilisées dans le traitement des herpèsvirus.
Shiraki et son équipe ont démontré que l'acide phosphonoacétique (PAA) inhiberait l'activité de l'ADN polymérase virale de l'HHV-6 (93).
La réplication virale de l'HHV-6 serait également inhibée par le PFA ou le GCV. Par contre, l'inhibition de la réplication virale n'a été observée qu'à des concentrations élevées d'ACV (94).
Le PAA, à des concentrations de 100 à 300 κg/mL, réduirait de façon significative la réplication virale. Il entraînerait une inhibition de la réplication de l'ADN viral alors que la réplication de l'ADN de la cellule hôte ne serait pas affectée, signifiant que seule l'ADN polymérase virale serait sensible au PFA. L'ACV à 20 κM ne semblerait pas interférer avec la production et le développement viral. L'ACV à 100 κM bloquerait seulement partiellement la réplication virale et l'ACV à 400 κM entraînait un arrêt complet de la réplication virale (55).
Dans une étude plus récente, Yoshida et son équipe ont évalué l'efficacité de plusieurs antiviraux sur la réplication de l'HHV-6A, l'HHV-6B et l'HHV-7. Les drogues étudiées étaient le PFA, l'ACV, le GCV, le penciclovir ou PCV, le CDV et son dérivé cyclique le cCDV. Des études de la concentration inhibitrice efficace à 50% (CI50) ont été réalisées à l'aide de méthodes de détections antigéniques par dot-blot (95) dans les cellules mononucléées de sang de cordon infectées par l'HHV-6A, l'HHV-6B et l'HHV-7 à une multiplicité d'infection de 0,004 CCID50/cellule. Les résultats de l'étude sont reportés dans le tableau 3. Les auteurs concluent de ces essais que le CDV et le cCDV sont les deux drogues les plus efficaces contre ces 3 virus et que l'ACV et le PCV sont les moins efficaces (96).

Il paraît assez normal que l'ACV soit moins efficace contre la réplication de l'HHV-6 car, ce virus est dépourvu du gène codant la thymidine kinase, enzyme spécifique nécessaire qui permet la première phosphorylation de l'ACV.
Une étude récente réalisée sur l'effet des sophocarpines a permis de conclure que ces drogues possédaient un bon potentiel d'inhibition de la réplication de l'HHV-6B et pourraient représenter un enjeux prometteur dans le développement de nouveaux traitements antiviraux moins agressifs (97).
1.10.3. Résistances développées par l'HHV-6 :
La sensibilité de l'HHV-6 aux antiviraux in vitro semble être similaire à celle du CMV puisque l'HHV-6 paraît être sensible au GCV, au CDV, au PFA qui sont les agents antiviraux les plus couramment utilisés pour le traitement des infections au CMV. L'infection active à l'HHV-6 est souvent associée à celle du CMV et bien que non spécifiquement traitée, elle se trouve, exposée au traitement anti-CMV. Cette exposition aux drogues antivirales entraîne la plupart du temps une inhibition de la réplication virale, mais peut aussi aboutir à la sélection de souches résistantes. Manichanh et ses collaborateurs ont mené une étude sur l'effet de l'exposition massive du GCV sur une souche en culture de l'HHV-6 (98). Ils ont également cherché des isolats de souches résistantes de l'HHV-6 chez des patients infectés par le CMV et traités au GCV pendant une longue période. Après isolement d'une souche de l'HHV-6 résistante au GCV en culture, et séquençage des gènes de l'ADN polymérase virale (U38) et du gène de la protéine thymidine kinase viral (U69), deux mutations ont été détectées qui sembleraient être associées à la résistance à l'anti-viral : la mutation A961V sur le gène U38 et la mutation M318V sur le gène viral U69. Par contre, seulement un seul patient (1/4) traité au GCV et infecté par l'HHV-6 semblait présenter la mutation M318V sur le gène U69 dans les cellules mononucléées du sang périphérique (PBMC). La mutation A961V n'a été recherchée que chez un seul de ces patients du fait du manque de PBMC chez les autres patients, elle n'a pas été détectée dans les PBMC. Les auteurs concluent de cette étude que la mutation M318V du gène U69 de l'HHV-6 est probablement associée à la résistance au GCV (98). La même équipe a également développé un système de PCR quantitative en temps réel suivant la technologie TaqMan permettant de déterminer facilement la sensibilité du virus face aux différents antiviraux utilisés, en quantifiant la multiplication du virus en contact ou pas avec différentes drogues (99).
Une étude récente a permis également de développer un système de baculovirus, permettant de tester les différentes mutations sur le gène viral U69 et de déterminer leurs conséquences sur la réplication virale au cours de traitements antiviraux. A l'aide de cette méthode, les auteurs ont identifié les mutations C448G et C463Y associées à la résistance au GCV (100).
1.11. HHV-6 et apoptose :
L'effet du virus de l'HHV-6 sur l'apoptose reste encore assez mal connu. Inoue et al. ont démontré plusieurs faits (101, 102) :
- L'inoculation seule par l'HHV-6 sans stimulation cytokinique exogène induit faiblement l'apoptose de cellules T CD4+ humaines de la lignée JJHAN.
- L'infection virale rend les lymphocytes T CD4+ plus sensibles à l'apoptose in vivo (102).
- Le degré d'apoptose des cellules T CD4+ inoculées par l'HHV-6 est augmenté par le TNFκ.
- Le nombre des sous-unités p55 du récepteur du TNF (important pour le message d'apoptose) exprimé dans les cellules JJHAN est augmenté après inoculation des cellules par l'HHV-6.
- Bien que l'inoculation par l'HHV-6 ne soit pas affectée pas l'expression de Fas, elle rend les cellules JJHAN plus sensible à l'apoptose sous stimulation par l'anticorps antiFas.
- L'apoptose apparaît préférentiellement dans les cellules négatives pour les antigènes de l'HHV-6.
- L'entrée de l'HHV-6 et sa réplication ne semblent pas requises pour l'induction de l'apoptose dans les cellules T.
- L'apoptose a lieu de façon prédominante dans les cellules non infectées par l'HHV-6 ou dans les cellules infectées par l'HHV-6 mais non productives.
Le mécanisme par lequel le virus induit l'apoptose dans les cellules non infectées reste inexpliqué. Les auteurs ont émis l'hypothèse qu'un composant de la structure du virus : la glycoprotéine 120 (gp120), ou certaines cytokines produites par les cellules infectées par l'HHV-6, seraient susceptibles de sensibiliser les cellules non infectées « bystander » à l'apoptose.
Ichimi et al. ont également montré que l'HHV-6 pouvait induire l'apoptose des cellules infectées après stimulation avec de l'interleukine-2 (103).
Mais Inoue et al. ont aussi proposé, de façon contradictoire, une hypothèse selon laquelle des produits de gènes viraux pourraient également inhiber l'apoptose (101).
On peut conclure de ces différentes études que l'implication de l'HHV-6 dans le processus apoptotique est encore très incertaine, trop de données sont à l'heure actuelle contradictoires. L'apoptose qui se manifeste suite à l'infection de lignées cellulaires par l'HHV-6 dans ces études semble liée, dans tous les cas, à des stimulations extérieures. Il est encore tout à fait incertain que le virus seul soit capable d'induire l'apoptose des cellules dans lesquelles il se trouve, ou celle des cellules non infectées situées à proximité.
2. Les lymphomes:
2.1. Les lymphomes non hodgkiniens :
2.1.1. Présentation générale :
Les lymphomes non hodgkiniens (LNH) sont des proliférations malignes du tissu lymphoïde. Les cancers du système lymphatique sont appelés lymphomes. Le système lymphatique aide l'organisme à se défendre contre les maladies. Il comprend un réseau de vaisseaux lymphatiques, longeant les artères, et les veines et reliant des ganglions lymphatiques situés au niveau du cou, de la région axillaire, du thorax, de l'abdomen et de la région inguinale (Figure 5). Les vaisseaux lymphatiques transportent la lymphe contenant les lymphocytes participant à la défense de l'organisme contre les infections. Le LNH apparaît généralement dans les lymphocytes présents dans l'un ou dans plusieurs ganglions lymphatiques. Les cellules anormales peuvent demeurer dans ces ganglions (LNH localisé) ou se propager à d'autres parties du système lymphatique (LNH généralisé).
Les LNH sont la conséquence d'une prolifération monoclonale de cellules B ou T bloquées à un stade de différenciation donné. Lors de la transformation maligne, le lymphocyte B ou T conserve la majeure partie des antigènes de différenciation et les critères morphologiques de la cellule dont il dérive. Les LNH regroupent des maladies tumorales lymphoïdes d'agressivité variable. Ils s'observent à tout âge, mais l'âge médian de survenue se situe entre 55 et 60 ans. L'homme est plus touché que la femme avec un rapport des sexes de l'ordre de 1,8. Les LNH sont les plus fréquentes des hémopathies malignes (7/100 000 habitants/an). Ils sont en moyenne trois fois à cinq fois plus fréquents que le lymphome de Hodgkin et ce dans tous les pays du monde (104). Ils représentent environ 85% de tous les cas de lymphomes. Les taux d'incidence des LNH les plus faibles sont observés dans les pays en voie de développement. Dans les pays occidentaux, les lymphomes B sont les plus fréquents (80% ou plus) alors que dans le sud du Japon par exemple, ce sont les lymphomes T qui prédominent (105). La fréquence des LNH est de 55 000 nouveaux cas par an aux USA.
2.1.2. Signes et symptômes du LNH :
Il existe un certain nombre de symptômes du LNH, mais aucun n'est vraiment spécifique car tous les symptômes associés au LNH ne sont pas nécessairement annonciateurs d'un LNH. Parmi les différents signes décrits, se trouvent des gonflements indolores des ganglions lymphatiques au niveau du cou, de la région axillaire, inguinale ou de l'abdomen. Des ganglions thoraciques peuvent entraîner des troubles de compression (toux, douleurs thoraciques). Il peut exister une altération de l'état général avec une perte de poids, la fièvre témoignant souvent une maladie disséminée.
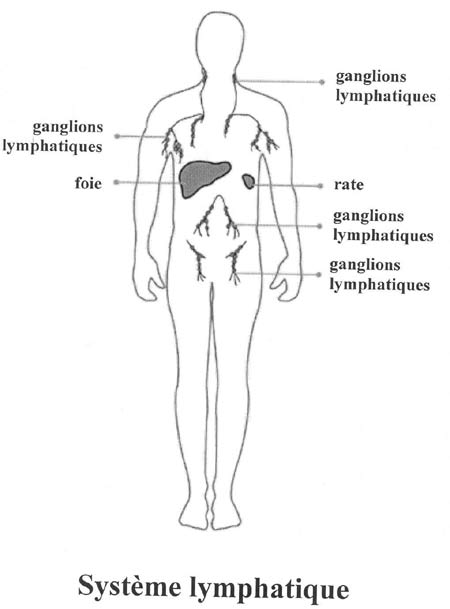
(Site internet : http://www.cancer.ca).
2.1.3. Diagnostic :
Le diagnostic des lymphomes malins est fondé sur la morphologie histologique et cytologique ainsi que par des moyens immunologiques d'identification. Le ganglion lymphatique est étudié après biopsie chirurgicale. La biopsie est indispensable au diagnostic. Elle montre la destruction de la structure histologique normale remplacée par les cellules lymphomateuses.
Les LNH de type B sont caractérisés par l'expression des antigènes de différenciation B (CD19, CD20, CD22...) et l'expression des immunoglobulines de surface ou cytoplasmiques. Les LNH de type T sont identifiés par des antigènes spécifiques des cellules T (CD2, CD3, CD7...), des antigènes de différenciation fonctionnelle (CD4, CD8). Des études cytogénétiques et moléculaires sont également effectuées sur la biopsie (106).
L'objectif du diagnostic des lymphomes malins est de situer la population tumorale dans le schéma de la physiologie du tissu lymphoïde normal. L'étude du compartiment fonctionnel lymphoïde B que le lymphome malin envahit initialement (centres folliculaires, zone du manteau ou marginale) permet de classer les lymphomes malins B en de nombreuses entités différentes. Les lymphomes malins observés en France sont de type immunologique B dans 85% des cas.
2.1.4. Classification des LNH :
La structure histologique de la tumeur permet de distinguer deux grands types de LNH :
Les lymphomes de structure nodulaire (ou folliculaire) dans lesquels une certaine persistance des follicules lymphoïdes existe. Il s'agit de prolifération de cellules B. Des sous-types ont été distingués en fonction essentiellement de la morphologie des cellules proliférantes.
Les lymphomes de structure diffuse dans lesquels le ganglion est complètement homogénéisé par une prolifération uniforme.
Les néoplasmes de cellules B comprennent les néoplasmes de cellules B précurseurs ainsi que les néoplasmes de cellules B matures. De même que les néoplasmes T et NK comprennent les néoplasmes de cellules T précurseurs et les néoplasmes de cellules T matures. Mais beaucoup de classifications de LNH ont été établies dans les 30 à 40 dernières années. L'ensemble des LNH classés est répertorié dans le tableau 4 (107, 108) suivant la dernière classification de l'Organisation mondiale de la santé (OMS) « World Health Organization classification of neoplasic diseases of the haematopoietic and lymphoid tissues ».
Après la confirmation du diagnostic, il faut déterminer le stade du cancer afin de choisir le traitement le plus adapté et le plus efficace. Le stade du cancer dépend de la taille de la tumeur et de son degré de propagation dans l'organisme.

OMS : Organisation Mondiale de la santé; « World Health Organization classification of neoplasic diseases of the haematopoietic and lymphoid tissues » (Harris et al., 2000).
2.1.5. Traitements :
Il est difficile de donner les grandes lignes des traitements dans les cas de LNH car chaque cas est particulier, les traitements actuellement employés reposent sur des combinaisons de chimiothérapies, de radiothérapies, de thérapies biologiques. On a également parfois recours à des greffes de moelle osseuse ou de cellules souches périphériques avec de bons résultats.
2.1.6. Biologie du LNH :
2.1.6.1. Causes biologiques :
Les LNH rassemblent des catégories très variées de lymphomes. C'est la raison pour laquelle il est difficile de discuter des causes probables du développement de la lymphomogenèse de l'ensemble de cette catégorie en généralisant les processus. Les LNH n'étant pas le sujet de notre travail de thèse, nous ne développerons pas les différentes étiologies des lymphomes non hodgkiniens du fait de la grande diversité des pathologies regroupées dans cette catégorie.
Citons juste, parmi les étiologies évoquées :
- des anomalies chromosomiques particulières très fréquentes, de type translocations, duplications ou délétions impliquant le plus souvent le chromosome 14 (109),
- des anomalies du système immunitaire.
2.1.6.2. Facteurs d'origine infectieuse :
L'infection par le virus EBV est très répandue dans les lymphomes. Le virus EBV infecte les lymphocytes B. En Afrique noire, 100% des cas de lymphome de Burkitt sont associés à l'EBV.
L'infection par le VIH est un facteur favorable à la survenue de LNH immunoblastiques et de type de Burkitt. Cinq pour cent des porteurs du VIH développent des lymphomes.
Le rôle du virus simien SV40 est un polyomavirus, qui infecte largement la population générale mais son implication dans la carcinogenèse chez l'homme est controversé. Des études récentes, montrant l'existence de séquences d'ADN de SV40 dans des lymphomes non hodgkiniens, relancent le débat
Des travaux ont permis d'étudier la présence des virus SV40, EBV et HHV-8 dans les ganglions de 154 patients atteints de LNH (76 étant positifs pour le VIH et 78 étant négatifs pour le VIH), de 54 patients atteints de cancer (colon, sein), 79 patients positifs pour le VIH et 107 négatifs pour le VIH sans lymphome. Des séquences du SV40 ont été détectées dans 42% des cas de LNH contre 0% chez les patients atteints de cancer ou dans la population de patients sans lymphome. L'ADN de l'EBV a été détecté dans 39% des cas de LNH VIH+ et 12% des cas de LNH VIH-. L'HHV-8 n'a jamais été détecté. Après analyse des lymphomes, le SV40 est retrouvé plus fréquemment dans les lymphomes diffus à grandes cellules (VIH+ ou -) et dans les lymphomes folliculaires observés chez les patients VIH-. Les auteurs concluent de cette étude que le rôle du virus SV40 doit être exploré et mieux défini pour préciser sa place en tant que cofacteur dans la survenue des LNH (110).
Deux autres études récentes ont également démontré la présence de séquences d'ADN génomique du SV40 dans 19% et 43% des patients atteints de LNH testés (111-113).
Mais l'implication du SV40 dans le développement des LNH reste controversée. MacKenzie et al. ont démontré, dans une étude plus récente, l'absence de séquences du virus SV40 dans les ganglions et dans le sang de 152 patients atteints de LNH en Grande Bretagne concluant qu'il était improbable que le SV40 ait un rôle dans la survenue des LNH (114).
2.2. Le lymphome de Hodgkin :
2.2.1. Généralités :
Le lymphome de Hodgkin (LH) est une prolifération maligne ganglionnaire caractérisée par la présence de cellules transformées de grande taille pathognomoniques, les cellules de Hodgkin et les cellules de Reed-Sternberg, (H/RS) (Figure 6). Les cellules de Reed-Sternberg (RS) sont indispensables au diagnostic, elles sont caractérisées par leur grande taille. Leur noyau est volumineux et bi ou multilobé avec en général un énorme nucléole. Les cellules de Hodgkin sont moins caractéristiques et insuffisantes à elles seules au diagnostic. Elles ont un seul noyau non lobé (115). Les cellules de H/RS représentent seulement une petite proportion (moins de 1%) de la population cellulaire dans le LH. La caractéristique du LH est, par conséquent, un petit nombre de cellules typiques de H/RS parmi un infiltrat cellulaire mixte (116, 117). Cet infiltrat cellulaire est principalement composé de lymphocytes T, B, de plasmocytes, d'histiocytes, d'éosinophiles et de neutrophiles mixtes (116, 117).
L'origine de cette cellule néoplasique de RS a été longtemps controversée. D'après des études basées sur les réarrangements des régions variables des immunoglobulines de cellules de H/RS micromanipulées provenant d'un infiltrat de tissus, la plupart des auteurs concluent à l'origine lymphocytaire de cette cellule et plus spécifiquement à son appartenance à la lignée B dans la plupart des cas de LH (118, 119). Le type cellulaire le plus abondant dans le tissu tumoral est la cellule T CD4+. Certaines d'entre elles sont associées intimement avec des cellules H/RS formant ainsi des rosettes autour d'elles. Après micromanipulation et étude des réarrangements des gènes bêta des régions variables des récepteurs des lymphocytes T (TCR), Roers et al. ont conclu que les cellules de H/RS attiraient les cellules T CD4+, mais de façon non spécifique (120). Ces cellules se trouvent au sein d'un environnement lymphocytaire pouvant former un granulome inflammatoire associé à un degré variable de fibrose.
Il existe 4 sous-types différents de lymphomes de Hodgkin déterminés par examen histologique de la biopsie :
- La forme scléro-nodulaire est la plus fréquente, regroupant environ 50 à 60% des cas. Elle est caractérisée par des zones de nodules séparées par des bandes sclérotiques (117).
- La forme à cellularité mixte regroupe 15% des cas. Elle ne présente pas de sclérose et montre une distribution cellulaire plus diffuse (117).
Dans ces 2 cas, l'infiltrat lymphocytaire est composé principalement de lymphocytes T. La forme avec déplétion lymphocytaire est plutôt rare (moins de 1% des cas) ; elle est caractérisée par une distribution diffuse des cellules avec une petite infiltration par des lymphocytes et présente des zones sclérosées et nécrotiques (117, 121).
- La forme avec déplétion lymphocytaire (ou riche en cellules tumorales) est très riche en cellules de Reed-Sternberg (117). Elle est assez rare et ne représente pas plus de 5% des cas.
- La forme à prédominance lymphocytaire montre une prédominance de lymphocytes B dans l'infiltrat cellulaire (122). Elle représente 5% des cas de LH. Dans ce sous-type, les cellules tumorales sont appelées cellules lymphocytiques et histiocytiques (L&H). Les cellules L&H ne sont pas aussi larges que les cellules de H/RS trouvées dans les LH classiques (117). Mais l'aspect général et surtout le phénotype particulier de ce sous-type conduisent à séparer cette affection du lymphome de Hodgkin classique et à en faire une catégorie à part entière (Recommandations OMS) (108).
Par conséquent, les formes scléro-nodulaire, à cellularité mixte et avec déplétion lymphocytaire sont regroupées sous la dénomination de lymphomes de Hodgkin classiques.
2.2.2. Epidémiologie :
Le lymphome de Hodgkin est une maladie plus fréquente chez l'homme que chez la femme avec un rapport homme/femme compris entre 1,5 et 2. Le LH est à l'origine de 0,24% de la mortalité par cancer en France. Dans les pays développés, il existe deux pics de la maladie, l'un survenant vers 30 ans et l'autre vers cinquante ans. Dans les pays en voie de développement, l'incidence du LH est plus réduite avec une prédominance chez l'enfant. En France, l'incidence est de 20 à 30 nouveaux cas de LH par an et par million d'habitants. Dans l'Union Européenne, le taux de mortalité lié au LH varie entre 0,5 et 1,7 pour 100 000 (Source : Fédération Nationale de Lutte Contre le Cancer). Le LH représente l'un des cancers les plus fréquents de l'adulte jeune. Même si le LH est peu fréquent, il ne peut être considéré comme une maladie rare.
2.2.3. Les manifestations cliniques :
Dans la majorité des cas, il s'agit d'un adulte jeune présentant une adénopathie périphérique isolée, indolore et non inflammatoire, le plus souvent cervicale ou sus-claviculaire, plus rarement médiastinale, découverte sur une radio de thorax systématique ou demandée devant des signes de compression (toux, douleurs thoraciques). Il peut s'agir d'un tableau de poly-adénopathies fébriles, voire d'une fièvre au long cours sans cause infectieuse retrouvée, avec altération de l'état général, amaigrissement...Ces formes sont souvent révélatrices chez le sujet âgé et témoignent d'une maladie d'emblée évoluée.
2.2.4. Diagnostic :
Le diagnostic du LH repose sur la mise en évidence des cellules de Reed-Sternberg au sein d'une réaction inflammatoire (Figure 6). La cytoponction ganglionnaire avec frottis a une valeur d'orientation. Elle peut montrer un frottis polymorphe constitué de cellules hyperbasophiles, de plasmocytes, d'éosinophiles, de petits lymphocytes mais les cellules de RS sont rarement retrouvées. La biopsie chirurgicale d'une adénopathie superficielle ou profonde est indispensable pour affirmer le diagnostic et préciser le type histologique. Le degré d'extension de la maladie permet de définir 4 stades cliniques. Le stade I consiste en une atteinte d'un seul groupe ganglionnaire ou de 2 territoires ganglionnaires contigus du même côté du diaphragme. Le stade II correspond à une atteinte de 2 territoires ganglionnaires non contigus mais du même côté du diaphragme. Le stade III est une atteinte de groupes ganglionnaires sus et sous-diaphragmatiques. Enfin le stade IV comporte une atteinte viscérale associée : foie, coeur, poumon, tube digestif, moelle, os, système nerveux. Cette classification en stades du lymphome de Hodgkin repose sur les modifications dites 'de Cotswolds' de la classification d'Ann Harbor de 1970. Elle oriente la thérapeutique et présente un intérêt pronostic. Le phénotype en immuno-marquage des cellules de RS est particulier : CD30+, CD15-, CD45- (marqueur T négatif) (115).
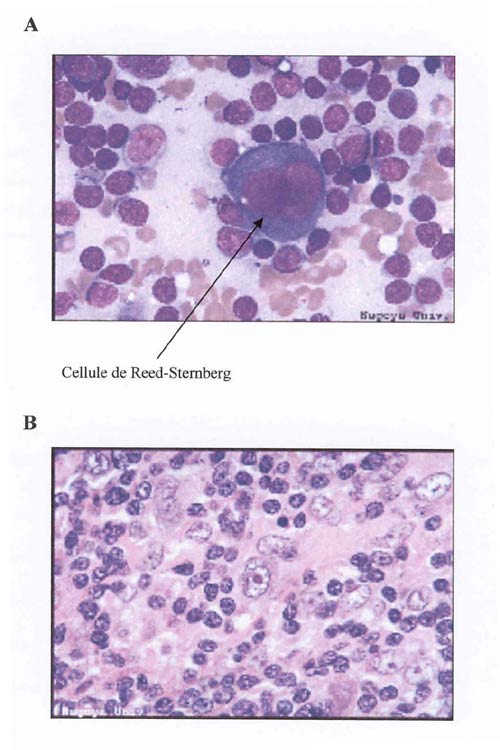
(A) La cellule de Reed-Sternberg est la cellule typique du lymphome de Hodgkin permettant de diagnostiquer la pathologie. (B) Photo de l'infiltrat cellulaire trouvé au niveau de ganglions de patients atteints de lymphome de Hodgkin. Photos provenant de la collection de l'Université de Nagoya (Site Internet http://www.nagoya-u.ac.jp/english/).
2.2.5. Traitements :
Le traitement repose en principe sur la radiothérapie dans les stades I et II et sur la chimiothérapie dans les stades III et IV, mais en pratique, les deux types de traitements sont souvent associés chez les patients. L'association de la chimiothérapie et de la radiothérapie permet maintenant de guérir près de 80 % des malades atteints de LH. Des efforts pour réduire la toxicité à long terme de ces traitements permettraient d'améliorer le pronostic de cette maladie.
2.2.6. Biologie du lymphome de Hodgkin :
Malgré le fait que le lymphome de Hodgkin ait été décrit il y a 171 ans par Thomas Hodgkin (123), cette pathologie reste encore énigmatique. De nombreuses dérégulations cellulaires ont cependant été mises en évidence (Figure 7) comme :
- des mutations de gènes cellulaires,
- des anomalies de profils d'expression de certains gènes cellulaires,
- des activations constitutives de facteurs cellulaires de transcription,
- des productions anormales de cytokines,
- des dérégulations provenant de différents virus (Figure 7).
Ces différentes dérégulations sont résumées dans la figure 7 et seront développées dans les prochains paragraphes.
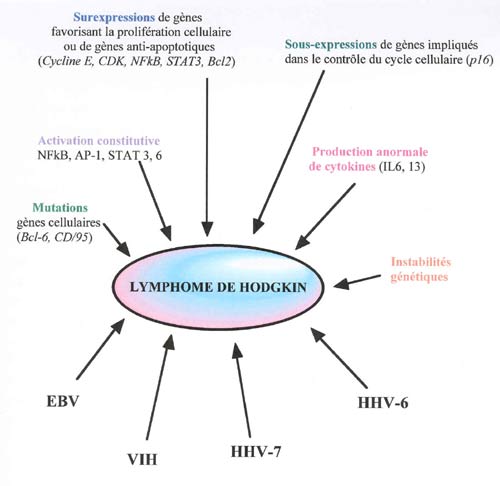
Il existe un certain nombre de dérégulations cellulaires et virales associées au LH. L'EBV semble être associé à un grand nombre de cas de lymphome de Hodgkin. Le VIH semble profiter d'un état d'immunodépression du patient. L'implication de l'HHV-7 paraît plus aléatoire et celle de l'HHV-6 est le sujet de ce travail.
2.2.6.1. Mutations au niveau de gènes dans les cellules de H/RS :
Certains auteurs ont mis en évidence des mutations au niveau de certains gènes cellulaires spécifiquement dans les cellules de H/RS (124-130). L'ensemble des mutations trouvées au niveau de gènes cellulaires dans les cellules de H/RS est répertorié dans le tableau 5.
Il est intéressant de noter l'absence de mutations sur le gène p53 et sur le gène ras qui sont normalement mutés dans un grand nombre de cancers humains et dont les mutations sont associées au phénotype transformant de la cellule tumorale. Ces deux gènes interviennent dans le contrôle de la prolifération des cellules humaines. Et si ces deux gènes sont associés au développement du LH, leur implication ne provient pas de mutations sur leur gène. Les différentes fonctions de la p53 seront abordées dans le prochain chapitre.
Par contre, des mutations de la partie 5' non codante du gène BCL6, considérées comme pouvant être associées à la pathogénicité de plusieurs lymphomes comme les lymphomes folliculaires (131, 132), sont détectées dans 100% des cas de lymphome de Hodgkin dans les cellules de H/RS dérivant de cellules B, d'après l'étude réalisée par Seit et al. (124). Par contre, aucune mutation n'a été détectée sur le gène BCL6 dans les cas de cellules H/RS dérivant de cellules T (124). Wlodarsk et al. ont également démontré une présence de réarrangement du gène BCL6 dans des cas de lymphome de Hodgkin du sous-type à prédominance lymphocytaire mais aucun dans les cas de lymphome de Hodgkin classiques (133). Les mutations présentes sur la région 5' non codante du gène BCL6 sont également fréquemment trouvées dans des cas de lymphomes B (134). La protéine BCL6 est un répresseur transcriptionnel intervenant dans la prolifération cellulaire et dans la différenciation (135). Les mutations au niveau de la région 5' non codante du gène BCL6 entraîneraient une dérégulation de l'expression de la protéine BCL6 (136). Mais les conséquences exactes de ces mutations ne sont pas clairement élucidées, elles dépendraient du type de cellules dans lesquelles elles se trouvent, du stade de différenciation cellulaire... (137). Kurosu et al. ont démontré qu'une surexpression de BCL6 entraînait une inhibition de l'apoptose dans le lymphome B (138).
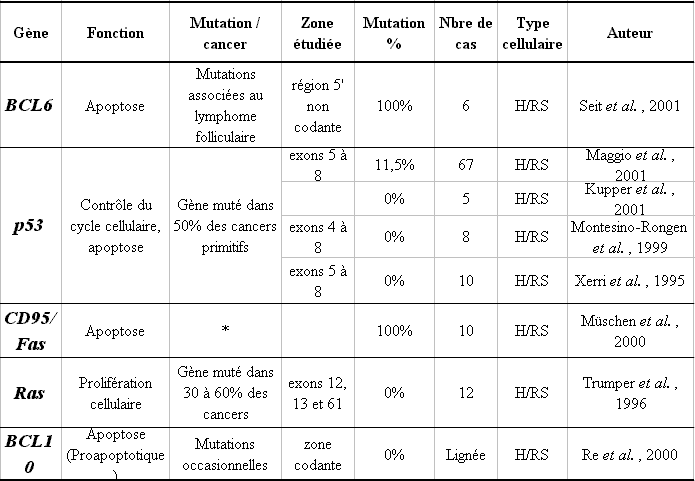
Le gène CD95/Fas semble également muté dans les cellules RS de 100% des patients testés atteints de LH (129). Par contre, les mutations trouvées au niveau de ce gène sont différentes les unes des autres. Trois mutations clonales à l'intérieur de la région 5' ont été détectées dans un cas. Deux mutations à l'intérieur du dernier exon codant le domaine de mort ont également été détectées dans les cellules de H/RS d'un autre cas, la moitié des cellules H/RS portaient des codons « stop » de façon monoallélique. Les tumeurs restantes présentaient des mutations monoalléliques entraînant le remplacement d'un acide aminé par un autre. Ces différentes mutations entraînent probablement des problèmes au niveau de la fonction de CD95. CD95/Fas est un récepteur de mort spécialisé dans l'induction de l'apoptose (139, 140). Une fois stimulé, il induit l'activation de la cascade des caspases aboutissant à la mort de la cellule. En conclusion, des mutations somatiques du gène CD95 existent dans une certaine proportion des cas de lymphomes de Hodgkin classiques pouvant expliquer en partie l'échappement des clones précurseurs des cellules de H/RS à l'apoptose (129).
Il peut exister dans certains cas de lymphomes de Hodgkin des instabilités chromosomiques dans les cellules de H/RS (141-143) (Tableau 6).

Il est difficile de savoir exactement quelles sont les conséquences de ces différentes altérations génétiques, mais il est évident que les amplifications géniques ont des répercutions importantes sur les gènes impliqués dans ce phénomène, entraînant certainement des surexpressions de ces gènes. Dans les cellules de RS présentant l'amplification 9p23-24, une augmentation du nombre de copie des séquences chromosomiques correspondantes au gène JAK2 a été détectée (143). JAK2 est une tyrosine kinase impliquée dans la transduction de différents signaux cellulaires, comme la prolifération, la différenciation ou l'apoptose. De même, Kupper et al. ont détecté dans les cellules de RS une amplification du gène HDM2 (inhibiteur de la p53) dans 4/6 (66,7%) des cas de patients atteints de LH, ce qui pourrait entraîner une augmentation de l'expression de la protéine HDM2 (126). Une surexpression de cette protéine implique une inhibition massive de la protéine p53 qui ne peut alors plus jouer son rôle de contrôle de la prolifération cellulaire, pouvant aboutir à une prolifération anarchique de la cellule.
2.2.6.2. Dérégulation de l'expression de gènes dans les cellules de H/RS :
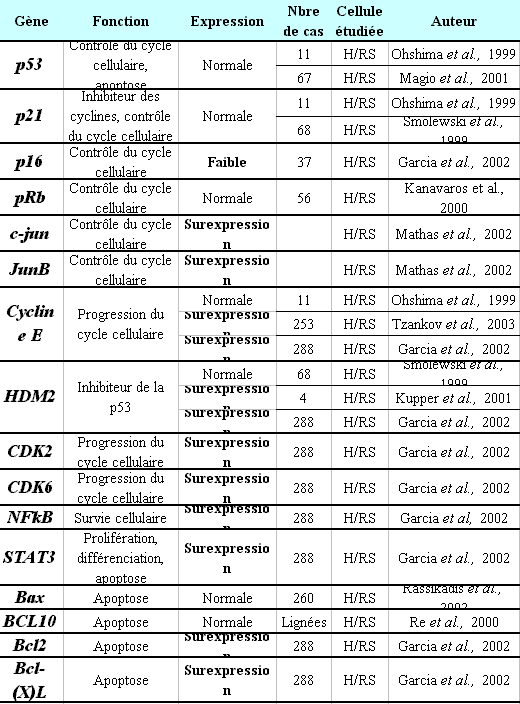
Il peut exister également des profils d'expression de gènes cellulaires plus ou moins normaux au cours de la lymphomogenèse du lymphome de Hodgkin (126, 144-153) (Tableau 7).
Au vu des résultats répertoriés dans le tableau 7, globalement, nous pouvons classer la dérégulation de l'expression des gènes dans les cellules de H/RS en deux catégories :
- les gènes inhibant la prolifération cellulaire comme c-jun et JunB. Ces deux gènes c-jun et JunB présentent une surexpression dans les cellules de H/RS (153). Ils sont considérés comme des régulateurs négatifs de la prolifération cellulaire. Par conséquent, ce phénomène aboutirait plutôt à un arrêt de la multiplication des cellules.
- les gènes favorisant la prolifération cellulaire. Certains auteurs ont détecté une surexpression des gènes de la cycline E (149, 150), HDM2 (126, 150), CDK2, CDK6, p50, p65, Bcl-2, Bcl-(X)L et STAT3 (150), et une expression plus faible du gène p16 (150). Les différentes dérégulations de l'expression de ces gènes aboutissent à une prolifération anarchique des cellules.
Plusieurs équipes ont démontré une expression plus faible de la protéine p16 dans les cellules de H/RS (154, 155). La protéine p16 appartient à la famille des inhibiteurs des cyclines ; elle inhibe spécifiquement le complexe cycline D1/CDK4 (cycline dépendante des kinases) (156, 157), kinase nécessaire à la progression du cycle cellulaire en phase S. Il existe des mutations fréquentes du gène de la p16 au cours des cancers (158), aboutissant à des divisions incontrôlées des cellules. L'expression faible de la protéine p16 détectée dans le LH pourrait provenir de l'hyperméthylation retrouvée au niveau de la zone promotrice du gène p16 dans les cellules de RS (150). La protéine p16 ne peut certainement pas, dans ces conditions, assurer correctement sa fonction de contrôle du cycle cellulaire, pouvant aboutir à une prolifération anarchique des cellules.
Les gènes Bcl-2 et Bcl-(X)L codent des protéines cellulaires antiapoptotiques (159), les protéines, cycline E, CDK2, CDK6, interviennent au niveau de la transition G1/S permettant le passage des cellules en phase S. Par conséquent, leur surexpression respective s'associe au processus de transformation cellulaire.
Garcia et al. ont analysé l'expression de 29 gènes impliqués dans la régulation du cycle cellulaire et dans l'apoptose, par immunohistochimie et hybridation in situ sur micro puce à ADN, des cellules de H/RS de 288 biopsies de patients atteints de lymphome de Hodgkin. Ils ont révélé des altérations multiples au niveau des voies de signalisation, au niveau des points de contrôle des transitions G1/S et G2/M du cycle cellulaire ainsi qu'au niveau de l'apoptose (152).
Il existe par conséquent, un certain nombre de dérégulations dans l'expression des gènes dans les cellules de H/RS pouvant faciliter l'apparition du phénotype tumoral.
2.2.6.3. Activations constitutives dans les cellules de H/RS :
En plus des dérégulations de l'expression des gènes trouvées dans les cellules de H/RS, de nombreux auteurs ont pu mettre en évidence des phénomènes d'activation constitutive de certaines protéines cellulaires, pouvant accentuer les processus de transformations cellulaires. Un certain nombre de facteurs cellulaires de transcription semble être constitutivement activé dans les cellules de H/RS comme les facteurs AP-1, certains facteurs appartenant à la famille de STAT (signal de transduction et d'activation de la transcription) et le complexe NFkB.
Il existe une activation constitutive du facteur de transcription AP-1 dans toutes les cellules tumorales de patients atteints de lymphome de Hodgkin testés par l'équipe de Mathas. Le facteur de transcription AP-1, formé par l'hétérodimérisation des protéines Fos et Jun, intervient de façon positive dans les processus de transduction du signal de multiplication cellulaire. AP-1 activé favorise la prolifération de la cellule de RS. Il coopère avec NFkB et stimule l'expression de la cycline D2 régulatrice du cycle cellulaire ainsi que du récepteur CCR7 et du proto-oncogène c-met dans les cellules primaires de RS. Les auteurs ont conclu à un rôle important du facteur AP-1 dans la lymphomogenèse du lymphome de Hodgkin (153).
L'équipe de Kube a démontré une activation constitutive du facteur de transcription STAT3 dans les cellules de H/RS de 5/7 des lignées cellulaires dérivées de LH testées (160). De même, une phosphorylation constitutive de STAT6 et STAT3 semble être un phénomène courant au niveau du LH. STAT 6 est phosphorylé constitutivement dans les cellules de RS de 5/5 des lignées dérivées de LH ainsi que dans les cellules de H/RS de 25/32 (78%) des cas de LH classiques (161). Les facteurs de transcription de la famille des STAT sont phosphorylés pour être actifs (162, 163). Ils interviennent dans la prolifération cellulaire, la différenciation et l'apoptose. Le facteur STAT3 est trouvé constitutivement actif dans certains processus de transformation cellulaire, au cours desquels il induit la prolifération cellulaire en accélérant la progression du stade G1/S dans le cancer du pancréas par exemple (164). L'activation de STAT3 est nécessaire à la transformation cellulaire (165). La voie de signalisation induite par STAT6 passe par l'IL-13, une cytokine fréquemment exprimée dans les cellules de H/RS du LH et la signalisation par l'IL13 est responsable de l'activation constitutive de STAT6 observée dans les cellules de H/RS, aboutissant à une boucle d'activation constitutive. Par contre, il n'y a pas de phosphorylation constitutive de STAT5 dans les cellules de RS dérivant de LH (161).
Dans de nombreux cancers primitifs ainsi que dans un certain nombre de lignées cellulaires tumorales, il existe une activation constitutive du facteur de transcription NFkB.
L'équipe de Bargou a démontré un niveau élevé d'activité du facteur NFkB dans des lignées cellulaires de H/RS dérivées de patients atteints de lymphome de Hodgkin et dans des cellules de H/RS isolées de fluide péricardique d'un patient atteint de LH. Bargou et al. ont montré également une présence constitutive nucléaire des sous-unités p50 et p65 de NFkB, ainsi qu'une activité constitutive du facteur de transcription hétérodimérique NFkB p50/p65 dans le noyau de ces cellules, alors que ces deux phénomènes sont normalement observés à des périodes limitées après diverses stimulations (166). Ils ont démontré aussi que l'activation constitutive de NFkB est nécessaire à la prolifération et à la survie des cellules tumorales de Hodgkin leur permettant d'échapper à l'apoptose suite à des conditions de stress. Leur étude permet, par conséquent, d'identifier NFkB comme un composant important dans la compréhension de la pathogenèse du LH (167).
Krappmann et al. ont démontré que dans certains cas de lymphome de Hodgkin, il existait une activité aberrante de l'inhibiteur de NFkB, IkB, pouvant expliquer l'activation constitutive de NFB (168).
Cabannes et al. ont détecté la présence de mutations sur le gène IkBa dans les cellules de H/RS de 2 lignées cellulaires dérivant d'un lymphome de Hodgkin ainsi que dans les cellules de H/RS de 2/8 (25%) des patients atteints de lymphome de Hodgkin pouvant expliquer l'activation constitutive de NFκB dans ces cellules (169).
Emmerich et al. ont également analysé le gène, l'ARNm et la protéine de l'inhibiteur de NFkB, IkBa dans des cellules primaires et en culture de H/RS. Dans des biopsies de patients atteints de lymphome de Hodgkin, une surexpression de l'ARNm d'IkBa était détectée dans les cellules de H/RS. Dans deux lignées cellulaires de H/RS (L428 et KM-H2), des mutations sur le gène IkBa étaient présentes, aboutissant à une protéine avec une extrémité C-terminale tronquée. De plus, des mutations monoalléliques étaient présentes dans des cellules de H/RS chez 1/10 (10%) des patients aboutissant à une protéine comparable à la précédente avec une extrémité C-terminale tronquée. Les auteurs ont conclu de cette étude que les mutations trouvées sur le gène IkBa contribuaient à l'activité constitutive de NFkB dans les cellules de H/RS en culture et primaires, et par conséquent, étaient impliquées dans la pathogénicité du lymphome de Hodgkin (170).
Les conséquences de l'activation constitutive du complexe NFkB ont été déterminées dans une étude récente. Hinz et al. ont déterminé les gènes cibles de NFkB comme étant des chimiokines, des cytokines, des récepteurs, des régulateurs de l'apoptose, des facteurs de transcription...NFkB est recruté directement sur le promoteur des gènes STAT5a, IL13 et CCR7 (Chemokine Receptor 7) permettant leur expression et leur activation permanente, causant une surexpression de STAT5a dans la plupart des cellules tumorales testées provenant de LH classique (171).
Les différentes dérégulations trouvées au niveau des cellules de Hodgkin et Reed-Sternberg du lymphome de Hodgkin participent à l'établissement du phénomène tumoral. Re et al. ont décrit que les lignées cellulaires de RS L1236 et L428 sont résistantes à l'apoptose induite par le ligand CD95 (ou ligand de Fas) bien que la protéine CD95 sauvage soit exprimée de façon normale (172).
Un certain nombre de dérégulations au niveau de l'expression et des fonctions des protéines cellulaires intervenant dans les contrôles du cycle cellulaire sont trouvées dans le lymphome de Hodgkin favorisant les processus de transformations cellulaires.
2.2.6.4. Dérégulations cytokiniques dans les cellules de H/RS :
Il existe une production aberrante de cytokines dans certains cancers pouvant être impliquée dans le développement et l'évolution du processus malin. Le lymphome de Hodgkin présente une production anormale de multiples cytokines (Tableau 8).
Nous n'avons pas établi une liste exhaustive de toutes les études réalisées sur le sujet, nous avons retenu certains travaux informatifs. Globalement, d'après ces différentes études réalisées sur la production de cytokines associées au LH, plusieurs points sont intéressants à noter :
Il n'y aurait pas de production d'IL3 et d'IL4, soit dans des lignées de cellules de H/RS soit dans les sérums de patients atteints de LH (173, 174).
Par contre, les interleukines IL1, IL2, IL5, IL6 et IL13 seraient produites avec des taux sériques plus élevés chez les patients atteints de LH que chez des sujets sains (173-182). Ces productions de cytokines seraient également détectées par recherche des ARNm de ces cytokines dans les cellules de H/RS de patients atteints de LH ou dans des lignées de cellules de RS dérivant de patients atteints de LH (173, 183).
On peut également noter que dans les cas des cytokines IL2, IL6 et IL13, les taux sériques élevés s'accompagnent mutuellement de l'expression de leur récepteur respectif au niveau de la cellule de H/RS, signifiant que le message induit par la cytokine peut être transduit dans la cellule (175, 182, 184).
Par conséquent, d'après un certain nombre d'auteurs, les productions anormales d'IL6, d'IL13 ainsi que d'autres cytokines pourraient être impliquées dans la biologie du lymphome de Hodgkin.
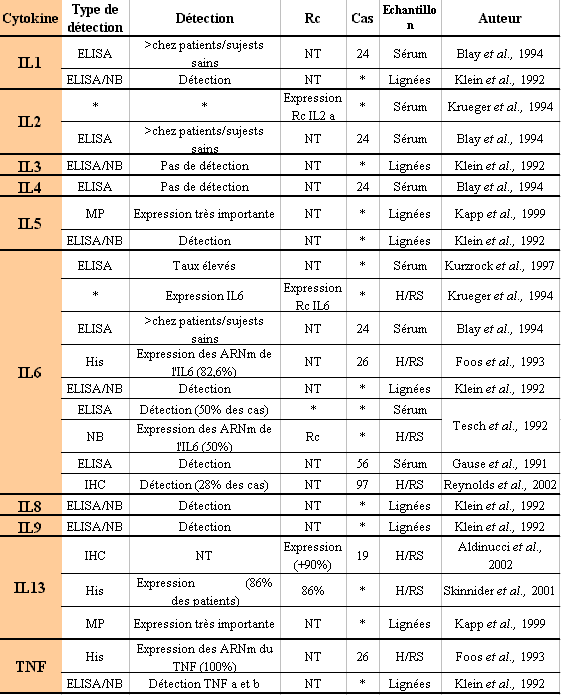
Il : interleukine, NB : northern blot, IHC : immunohistochimie, His : hybridation in situ, MP : micro-puce à ADN.
2.2.6.5. Implications virales dans le lymphome de Hodgkin :
2.2.6.5.1. Le virus d'Epstein Barr :
La présence du virus Epstein Barr (EBV) semble être associée au lymphome de Hodgkin dans 40 à 50% des cas dans les pays industrialisés. Nous avons regroupé les résultats obtenus pour quelques études dans le tableau 9 (78, 185-197).
Un certain nombre de données découle de ces différentes études :
- La présence du virus semble être variable selon le sous-type de la pathologie. Le sous-type à cellularité mixte présenterait plus fréquemment des séquences virales (80%) que le sous-type à déplétion lymphocytaire (71,4%) ou que le scléro-nodulaire (50%) (189, 198).
- A priori, la présence de ce virus ne semblerait pas affecter la survie des patients. Néanmoins, ceux atteints de LH et non infectés par l'EBV, présenteraient un meilleur pronostic surtout s'il s'agit d'hommes et de jeunes adultes (190).
- La présence du virus d'EBV chez des patients atteints de lymphome de Hodgkin pourrait également être liée à l'âge. Jarrett et al. ont démontré que le virus EBV semblait plus présent chez des patients très jeunes (enfants) et chez les personnes de plus de 50 ans (199). Par conséquent, plusieurs auteurs émettent l'hypothèse que l'EBV serait directement associé à certains cas de lymphome de Hodgkin.
- Le sous-type A du virus d'EBV semble être présent dans la majorité des cas de patients infectés dans la cellule de H/RS (191, 200). Néanmoins, le type B du virus d'EBV est également trouvé associé à certains cas de lymphome de Hodgkin chez les patients immunodéprimés (201).
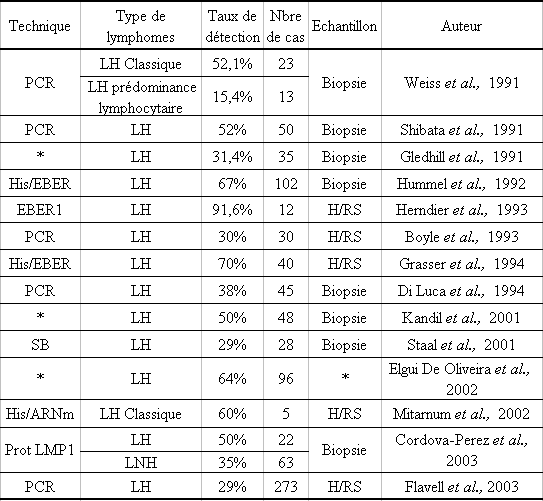
His : hybridation in situ, SB : southern blot, EBER : Epstein-Barr virus small RNA. LMP1 : protéine latente de membrane.
Finalement, Stachura et al. ont rapporté, dans une étude récente, le cas intéressant d'un patient atteint d'un LH pulmonaire présentant une co-infection à l'EBV et au CMV, ce qui est, a priori, assez rare (202).
2.2.6.5.2. Le virus de l'immunodéficience humaine (VIH) :
Plusieurs équipes ont rapporté un certain nombre de cas de LH associés au VIH (Tableau 10) (203-206).

Plusieurs conclusions ont été émises en ce qui concerne l'association de ce virus dans le LH :
Le risque de développer un LH serait majoré de 5 à 8 fois chez les sujets positifs pour le VIH avec une incidence plus importante du LH pendant la phase SIDA (207).
Le développement du LH (surtout les sous-types à cellularité mixte et à déplétion lymphocytaire) serait vraisemblablement influencé par l'immunosuppression présente chez les patients infectés (208).
La stimulation par le VIH du développement du LH semble être différent suivant les patients (203, 209). Par conséquent, certains auteurs proposent un dépistage automatique du VIH chez les patients atteints de LH (209).
Le LH associé au VIH serait de nature atypique (à cause du virus) avec un profil clinico-pathologique particulier du LH chez les patients infectés avec un nombre élevé de type à cellularité mixte et une fréquence importante des stades avancés du lymphome (206).
Les stades avancés de LH souvent trouvés chez les patients infectés impliquent souvent un pronostic de survie plus mauvais que chez des patients non infectés. Néanmoins, Diwan et al. ont présenté le cas d'une patiente de 38 ans avec un LH diagnostiqué en 1990 et infectée par le VIH. Cette patiente semblait en rémission complète 9 ans après le diagnostic initial (210). Cela prouve qu'une survie prolongée de patients infectés par le VIH et atteints de LH avec un pronostic vital faible est possible, même si elle est inhabituelle.
La survie des patients atteints de LH et infectés par le VIH serait améliorée depuis l'introduction des thérapies anti-rétrovirales hautement actives (211). Mais il semble, a priori, difficile de savoir si les risque de développer un LH est diminué depuis l'introduction des thérapies anti-rétrovirales (212).
2.2.6.5.3. Autres virus :
La présence de plusieurs autres virus est également signalée dans cette pathologie (Tableau 11), mais leur implication semble moins plausible (213-217).
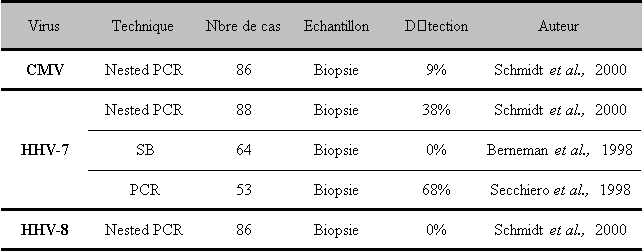
La présence des virus CMV, HHV-7 et HHV-8 semble être aléatoire chez les patients atteints de LH. Les virus CMV et HHV-8 sont rarement trouvés associés au LH, laissant suggérer une absence de corrélation entre ces virus et le développement de cette pathologie (214). Par contre, le virus HHV-7 est détecté dans un nombre de cas plus important (38% et 68%) indépendamment du sous-type de LH (213, 215), mais Secchiero et al. considéraient que le virus ne paraissait pas être impliqué directement dans la lymphomogenèse car aucune séquence d'ADN viral n'avait été détectée dans les cellules de Reed Sternberg, dans les cellules de Hodgkin et assez rarement dans les petits lymphocytes T (215).
Récemment Benharroch et son équipe ont apporté des preuves préliminaires suggérant une association possible entre le virus de la rougeole et le LH (216).
2.2.6.5.4. Implication de l'HHV-6 dans le lymphome de Hodgkin :
Détection du virus :
HHV-6 pourrait jouer un rôle étiologique dans la survenue du lymphome de Hodgkin. Différentes études ont pu mettre en évidence la présence de l'HHV-6 chez les patients atteints de LH :
En effet, de nombreuses études sérologiques ont estimé une prévalence élevée des IgG anti HHV-6 chez des patients atteints de désordres lymphoprolifératifs. L'équipe de Biberfeld a estimé cette prévalence à 50-70% des cas de patients atteints de lymphomes malins, 41% des patients avec une sarcoidosis, et 36% des patients atteints de syndrome de Sjogren. (218). L'étude menée par Shavanas et al. montrait une prévalence des IgG anti HHV-6 chez 95% des patients atteints de lymphome de Hodgkin contre 76% dans la population contrôle composée de sujets sains (219).
Torelli et al. ont montré que le titre d'anticorps anti HHV-6 était plus élevé chez les patients atteints de lymphome comparés aux sujets sains, mais la différence semblait être plus significative lorsqu'il comparait les patients atteints de lymphome de Hodgkin et les sujets sains (79).
Rojo et al. ont décrit la présence d'antigènes de l'HHV-6 dans les différents cas de lymphome de Hodgkin étudiés (220). Krueger et al. ont également trouvé la présence d'antigènes de l'HHV-6 dans 77,3% des patients atteints de lymphome de Hodgkin (175).
Des séquences d'ADN viral ont également été détectées par PCR et par hybridation in situ dans les adénopathies d'un grand nombre de patients atteints de lymphome de Hodgkin (Tableau 12) (75, 76, 78, 79, 221-223).
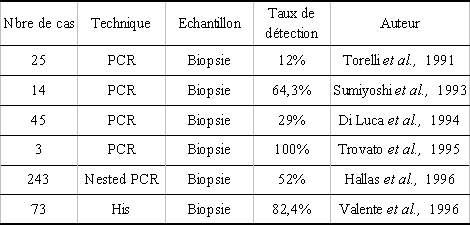
Plusieurs conclusions ont été émises à la suite de ces différentes études quant à l'association entre l'HHV-6 et le LH :
Dans la plupart de ces études, les séquences d'ADN viral ont été recherchées sur des extraits d'ADN de biopsies présentant un ensemble de types cellulaires différents. Mais des études ont été réalisées sur des populations cellulaires séparées et la localisation de l'HHV-6 semble controversée. Certains auteurs ont montré une absence de séquences virales dans les cellules de Reed-Sternberg et dans les cellules de Hodgkin (222, 224). Néanmoins, Krueger et al. ont révélé la présence de l'HHV-6 dans les cellules monocytiques incluant les cellules de Hodgkin et Reed-Sternberg plutôt que dans les cellules lymphoïdes (175).
Dans la plupart des cas de patients atteints de lymphome de Hodgkin, le type B de l'HHV-6 est majoritairement présent (92,3%, 94%) (78, 222).
De plus, dans la plupart des études, le virus HHV-6 est majoritairement trouvé dans le sous type scléro-nodulaire du LH. Di luca et al. ont détecté la présence du virus dans 69,2% des cas de LH à sous-type scléro-nodulaire et dans 23% pour les sous-types à cellularité mixte (78). Valente et al. ont trouvé des séquences d'ADN de l'HHV-6 dans respectivement 73,1%, 50% et 70% des sous-types scléro-nodulaire, interfolliculaire et à cellularité mixte (222).
Le virus de l'HHV-6 est plus souvent détecté chez des patients atteints de LH relativement jeunes. Hallas et al. ont démontré la présence de séquences d'ADN viral dans un grand pourcentage de patients ayant moins de 60 ans (54%) plutôt que chez des patients plus âgés (35%) (75).
Co-infection EBV et HHV-6 :
Une co-infection EBV/HHV-6 est souvent trouvée dans le lymphome de Hodgkin. Valente et al. ont démontré la présence simultanée des deux virus dans 54,9% des adénopathies de patients atteints de lymphome de Hodgkin (222). L'HHV-6 aurait la capacité de transactiver l'EBV. Flamand et son équipe ont décrit que le cycle réplicatif de l'EBV est activé par l'HHV-6. L'infection par l'HHV-6 entraînait une augmentation d'un facteur 10 de l'expression de l'antigène très précoce Zébra et des antigènes précoces EA-D et EA-R dans des lignées cellulaires productrices et non productrices de l'EBV. De même l'expression des produits de gènes tardifs appelés antigènes viraux de capside et glycoprotéine membranaire virale gp350 de l'EBV était aussi augmentée dans les lignées cellulaires productrices de l'EBV suite à l'infection par l'HHV-6 (225). Cuomo et al. ont démontré que l'infection par l'HHV-6 pouvait activer l'expression des gènes LMP-1 et EBNA-2 de l'EBV dans des lignées cellulaires EBV positives (Akata et P3HR-3), l'expression de ces deux gènes étant essentielle à l'immortalisation des lymphocytes B par l'EBV. Les auteurs ont conclu à un nouvel aspect de l'interaction entre ces deux herpèsvirus (226). Dans une étude plus récente, Flamand et al. ont décrit que le virus HHV-6 transactivait la transcription du gène Zebra de l'EBV à l'aide de l'élément de réponse à CREB (CRE) situé à l'intérieur du promoteur Zebra (227).
Par conséquent, l'activation de la réplication de l'EBV par l'HHV-6 pourrait contribuer à augmenter la pathogénicité de l'EBV ou les phénomènes d'immortalisation associés à l'EBV.
HHV-6 et lymphomogenèse :
L'implication de l'HHV-6 dans la lymphomogenèse du lymphome de Hodgkin reste encore incertaine. Plusieurs études ont démontré un caractère oncogénique de l'HHV-6.
Le génome entier du virus HHV-6A souche GS est capable de transformer des fibroblastes murins NIH3T3 (228), ou des kératinocytes humains RHEK-1 (229). Ces deux types de cellules transformées induisent un développement rapide de tumeurs dans des souris nude après transfection (228, 229). Le fragment sal-IL (Figure 8) du génome de l'HHV-6A (souche U1102) situé au niveau des DR présente une activité transformante (230). Après séquençage de ce fragment, l'équipe de Kashanchi a déterminé la présence de 7 cadres de lectures (ORF : Open Reading Frame) (48). Le premier (ORF1), correspondant au gène DR7, a la capacité de transactiver in vitro les LTR (Long Terminal Repeat) du VIH aboutissant à une augmentation de la production virale (48). Il est également responsable de la propriété transformante du fragment sal-IL (47). Les cellules exprimant la protéine DR7 sont tumorigènes quand elles sont injectées dans des souris nude alors que des cellules produisant une protéine tronquée ne le sont pas (47). De plus, Kashanchi et al. ont aussi démontré que la protéine DR7 de l'HHV-6A (souche U1102) a la capacité de se lier in vitro à la protéine p53 humaine sur sa zone de liaison à l'ADN, inhibant ainsi sa fonction de facteur de transcription et la rendant inactive (47).
Le gène viral DR7 du type A (souche U1102) mesure 1092 bases et est présent sous l'état de deux copies par génome. Il code une protéine de 363 acides aminés possédant des domaines hydrophobes, et présentant des homologies de séquences sur 3 domaines d'environ 15 acides aminés chacun avec deux protéines transactivatrices du CMV : TRS1 et IRS1 (appartenant à la famille US22). Les ARN messagers de DR7 sont détectés à partir de 18h jusqu'à 48h après l'infection par le virus HHV-6A souche U1102 des cellules HSB2. La protéine DR7 est détectée environ 18h jusqu'à 72h après l'infection. Kashanchi et al. ont montré dans leur étude, une transcription et une traduction simultanées de cette protéine DR7 (47). La fonction de la protéine DR7 est encore mal connue au sein du cycle viral, elle semble jouer un rôle de transactivateur de l'expression des gènes viraux mais aussi cellulaires.
DR7 n'est pas le seul gène candidat susceptible de jouer un rôle dans l'induction de la transformation cellulaire, il existe un certain nombre d'autres transactivateurs viraux potentiels plus ou moins bien connus. Ils appartiennent souvent à la famille US22 pour leur homologie de séquences avec les gènes US22 du CMV.
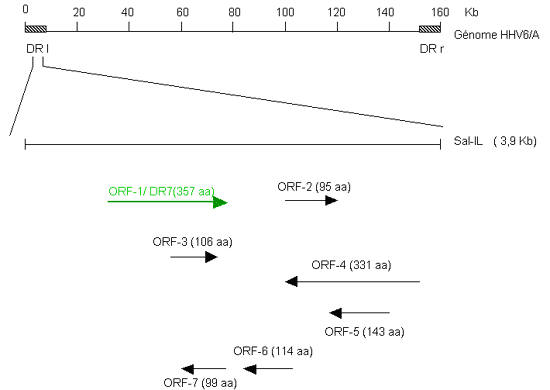
Localisation du fragment Sal-IL au niveau des Direct Repeat (DR) sur le génome de l'HHV-6A et localisation de l'oncogène viral DR7, également appelé ORF1 (Thompson et al., Virology, 1994 ; Kashanchi et al., Oncogene, 1997).
3. Proliferation cellulaire et deregulations
3.1. INTRODUCTION GENERALE :
Le processus de transformation cellulaire aboutissant à la formation tumorale est un processus très complexe dont les étiologies sont encore mal connues. L'apparition d'une transformation cellulaire implique la survie de la cellule mais avec des altérations de croissance causée par la dérégulation du cycle cellulaire.
Un certain nombre de virus à ARN ou ADN sont impliqués dans l'apparition de ces phénomènes de cancérisation. Ce travail de thèse portant exclusivement sur un membre de la famille des herpèsvirus humain, nous nous intéresserons dans ce chapitre aux virus à ADN et à leurs implications dans les processus de transformation cellulaire.
Les mécanismes utilisés par les virus à ADN dans les processus de transformation des cellules peuvent être très différents :
Altération du système immunitaire des cellules, pouvant limiter l'élimination des cellules tumorales (virus de la maladie de Marek, cytomégalovirus).
Insertion du génome viral au sein d'un chromosome cellulaire pouvant entraîner des dérégulations de l'expression des gènes cellulaires (papillomavirus).
Dérégulations du cycle cellulaire par inhibition de protéines essentielles aux différentes étapes de contrôle comme la p53, pRb ou la p21, souvent par interaction avec des protéines virales (papillomavirus, SV40, adénovirus, EBV, cytomégalovirus...) (Figure 15).
Altérations des signaux apoptotiques. Certains virus ont la capacité de suractiver le facteur de transcription NFκB responsable de messages de survie cellulaire, bloquant l'apoptose (EBV, CMV...).
Il s'agit d'une partie seulement des différentes dérégulations provoquées par des virus à ADN pouvant intervenir dans les processus de transformation cellulaire. Au cours de ce travail de thèse, nous nous sommes intéressés à la protéine p53 ainsi qu'au complexe NFκB, c'est la raison pour laquelle nous n'avons détaillé que ces deux aspects dans ce chapitre.
3.2. RAPPELS SUR LE CYCLE CELLULAIRE :
3.2.1. Introduction :
On appelle cycle cellulaire l'intervalle entre chaque division cellulaire, composée de 4 phases successives (Figure 9) : la phase G1 (pour Gap ou Growth phase 1), la phase S (DNA synthesis), la phase G2 (pour Gap ou Growth phase 2) et la phase M (pour mitose ou méiose). La phase G0 est l'état de repos des cellules qui ne se divisent pas.
Entre ces différentes étapes, des points de contrôle ou « check point » existent, ayant pour but de vérifier l'intégralité de la transmission de l'ADN de la cellule mère vers les cellules filles.
La succession des événements qui aboutit à la division cellulaire est en grande partie déterminée par l'expression séquentielle des cyclines. Elles exercent leur action grâce à l'association et l'activation de kinases dépendantes des cyclines (CDK). Les cyclines sont exprimées de façon cyclique (comme leur nom l'indique) (sauf la cycline D) alors que leurs partenaires CDK sont toujours présentes. Les CDK sont des petites protéines formant le domaine catalytique. Leur activation dépend de l'association avec les cyclines mais aussi de la phosphorylation sur un résidu thréonine très conservé.
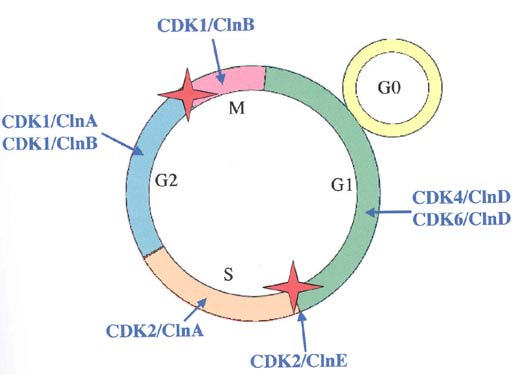
: points de contrôle
3.2.2. La phase G1 :
Pendant la phase G1, les membres de la famille de protéines «pocket» (pRb, p107 et p130) séquestrent par liaison le facteur de transcription E2F (231), le rendant inactif. E2F appartient à la famille des facteurs de transcription E2Fs, responsables de la régulation de l'expression des gènes impliqués dans la différenciation, le développement, la prolifération et l'apoptose (232). Le complexe pRb-E2F réprime l'expression des gènes du cycle cellulaire et ceci constitue un événement essentiel de régulation de la prolifération cellulaire. A l'approche de la transition G1/S, des cyclines de la phase G1 vont inactiver les membres de la famille de la pRb par phosphorylation. La phosphorylation de la pRb entraîne un changement de conformation de cette dernière libérant ainsi les facteurs de transcription E2F. E2F ainsi libérés se lient à l'ADN en association avec un autre facteur de transcription DP-1 et ensemble, ils induisent l'expression des cyclines E et A nécessaires pour le passage à travers la phase G1 (Figure 9 et 10).
Mais aussi, les facteurs de la famille «pocket» régulent la transcription des gènes impliqués dans la réplication de l'ADN et dans l'induction de l'expression des histones 2A et 2B, indispensables pour l'assemblage de l'ADN dans les nucléosomes.
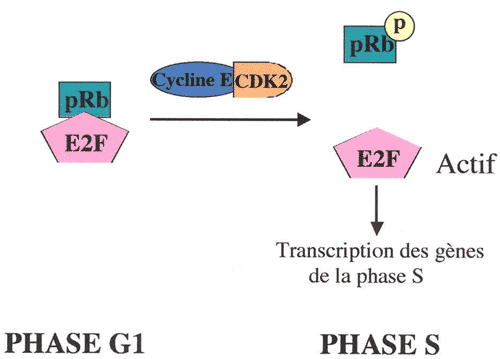
3.2.3. La phase S
Dans cette phase, l'action de la cycline A domine. Le complexe cyclineA/CDK2 agit avec les membres de la famille E2F et en parallèle avec les cyclines E/CDK2 pour réguler l'expression des gènes nécessaires pour la réplication de l'ADN (Figure 9).
3.2.4. La phase G2
Enfin, les cyclines de type B entrent en jeu et s'associent avec CDK1 (aussi appelé Cdc2). Le complexe cycline B/CDK1 avance le cycle dans G2 et prépare la cellule pour la mitose. Le passage en G2 est strictement contrôlé par l'activité de CDK1 qui est à son tour étroitement régulée (Figure 9).
3.2.5. La mitose
L'activité du complexe cyclineB/CDK1 permet la condensation de l'ADN (pour obtenir les chromosomes condensés caractéristiques, les chromatides, de la métaphase), le désassemblage de l'enveloppe nucléaire (dépolymérisation de la lamine) et la réarrangement du cytosquelette de tubuline (Figure 9).
3.2.6. Les points de contrôle du cycle cellulaire
Les principaux points de contrôle décrits sont :
- Un point de contrôle entre les phases G1 et S, qui autorise ou non la poursuite de la duplication de l'ADN et dont le régulateur majeur est la protéine p53.
- Un point de contrôle entre les phases G2 et M, qui autorise la division cellulaire, mais dont on connaît moins bien les protéines régulatrices (Figure 9).
Au cours de ces points de contrôle, des protéines cellulaires interviennent afin de vérifier si la cellule peut poursuivre son évolution dans le cycle cellulaire. Parmi ces protéines, il existe les inhibiteurs des CDK.
3.2.7. Les inhibiteurs des CDK
L'activité des CDK est réglée non seulement par le niveau d'expression des cyclines et par leur phosphorylation mais aussi par la présence d'inhibiteurs, les CDKI, présentant deux familles, la famille INK4 et la famille KIP/CIP.
3.2.7.1. La famille INK4
Les membres de la famille INK4 sont p15INK4B, p16INK4A, p18INK4C, et p19INK4D. Ils inhibent la progression dans le cycle cellulaire seulement en présence d'une protéine pRb fonctionnelle. Ils se lient spécifiquement aux kinases CDK4 et CDK6, qu'ils inhibent en empêchant la liaison de la cycline D. Il apparaît donc que ces inhibiteurs agissent pour empêcher les phosphorylations de la pRb en inhibant les kinases CDK4 et CDK6. La p15INK4B et la p16INK4A ont donc un rôle important en empêchant le déclenchement de la prolifération cellulaire et sont de ce fait des gènes suppresseurs de tumeurs, fréquemment altérés dans les tumeurs.
3.2.7.2. La famille KIP/CIP
Les membres de cette famille sont p21WAF1/CIP1, p27KIP1, p57KIP2. Ils se lient et inhibent la plupart des complexes cyclines/CDK. La p27KIP1 est une gène suppresseur de tumeur qui empêche la progression cellulaire en phase S en bloquant les complexes cycline E/CDK2 et cycline A/CDK2. La p21WAF1/CIP1 a une spécificité plus large puisqu'elle inhibe l'activité des complexes cycline E-A/CDK2 et cycline B/CDK1. Il s'agit de l'un des principaux médiateurs de l'arrêt du cycle cellulaire en réponse à divers stress cellulaires.
3.3. RAPPELS SUR P53 :
3.3.1. Généralités :
La p53 est une protéine cellulaire présentant un rôle capital au niveau des points de contrôle des phases G1/S et G2/M du cycle cellulaire. La protéine p53 est un facteur de transcription permettant l'activation de la transcription de gènes très variés, impliqués dans l'arrêt du cycle cellulaire, la réparation de l'ADN après dommage et dans l'apoptose (Figure 11). Elle est souvent présentée comme le « gardien du génome » (233).
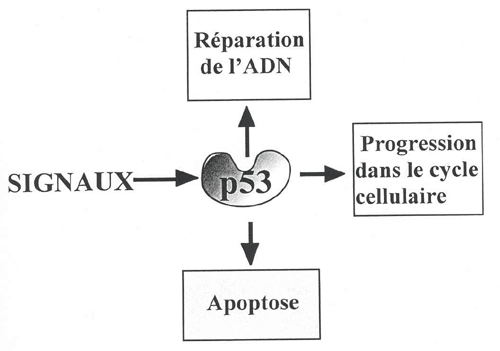
La protéine p53 humaine est une phosphoprotéine de 53 kDa codée par un gène de 20 kb contenant 11 exons (234), localisé sur le chromosome 17q13 (235). Elle appartient à une famille de gènes hautement conservés contenant au moins deux autres membres : p51/p63 (236, 237) et p73 (238). La protéine p53, composée de 393 acides aminés, comprend plusieurs domaines différents : un domaine amino-terminal nécessaire à la transactivation de la transcription (DTA), une région centrale spécifique de la liaison à l'ADN (DLA), une région linker flexible (RLF), un domaine de tétramérisation (DT) et un domaine de régulation (DR) hautement basique en position carboxy-terminale (239) (Figure 12).
Dans la cellule normale, la protéine p53 est présente en faible quantité, souvent liée à son principal inhibiteur HDM2 (Human Double Minute 2). En réponse à un stress cellulaire (hypoxie, exposition aux UV...) endommageant l'ADN, le niveau d'expression et l'activité de la protéine p53 augmentent. L'activation de la protéine p53 résulte de plusieurs mécanismes différents. Elle subit une série de modifications post-traductionnelles basées sur des phosphorylations et des acétylations. Puis elle s'associe en homotétramère qui paraît être sa forme la plus active (240). Enfin la protéine ainsi activée va être transloquée dans le noyau de la cellule où elle pourra exercer ses fonctions de facteurs de transcription. Suite à un stress cellulaire, la protéine p53 va, dans un premier temps, entraîner un blocage du cycle cellulaire aux transitions G1/S ou G2/M, puis, dans un deuxième temps, induire la réparation de l'ADN et finalement déclencher l'apoptose de la cellule si les dommages de l'ADN sont trop importants.
3.3.2. Rôles de la protéine p53 :
3.3.2.1. La protéine p53 et l'arrêt du cycle cellulaire :
3.3.2.1.1. Transition G1/S :
Pendant la phase G1, suite à un stress cellulaire, la protéine p53 augmente l'expression de la protéine p21Waf1/Cip1 qui inhibe les CDK, empêchant ainsi la libération de E2F et le passage à la phase S (Figure 13). La protéine p21 va, dans un deuxième temps, également inhiber le PCNA qui est un facteur auxiliaire des sous-unités κ et κ de la polymérase servant à faciliter l'accrochage de la polymérase sur l'ADN (241).
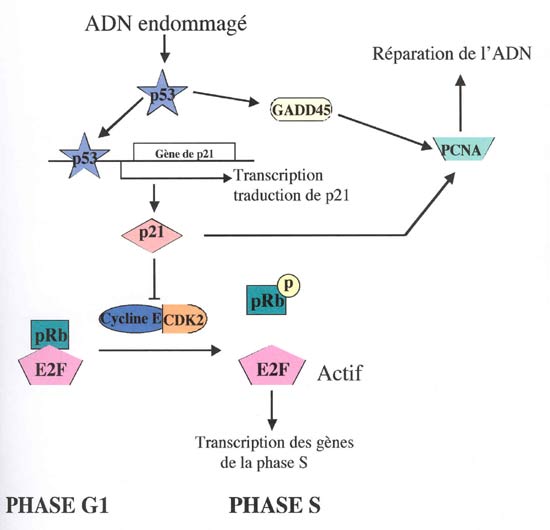
3.3.2.1.2. Transition G2/M :
En ce qui concerne la phase G2 (Figure 14), le mécanisme employé par la protéine p53 pour bloquer le cycle cellulaire est un peu différent ; il implique l'inhibition de la protéine cdc2 qui est une cycline dépendante des kinases nécessaire à l'entrée de la cellule en mitose (242). Dans la cellule normale, cdc2 est associée à la cycline B (243). A la fin de la phase G2, le complexe cdc2/cycline B est activé par phosphorylation permettant ainsi la progression de la cellule en mitose. Dans la cellule ayant subi des lésions de l'ADN, la protéine p53 va inhiber cdc2 par au moins 3 mécanismes différents :
Dans un premier temps, la protéine cdc2 va être inhibée par 3 protéines, GADD45, p21Waf1/Cip1 et 14.3.3s, dont les transcriptions sont induites par la protéine p53 (244). GADD45 inhibe le complexe cdc2/cycline B en se fixant sur cdc2 (245). P21Waf1/Cip1 est un inhibiteur universel des CDK, essentiel pour le maintien du contrôle G2/M (246). 14.3.3s se fixe à cdc2 et l'empêche de phosphoryler les protéines nécessaires à l'entrée de la cellule en mitose (247).
Dans un deuxième temps, la p53 va réguler cdc2 directement en se fixant sur la sous-unité catalytique du complexe cdc2/cycline B inhibant ainsi son activité de kinase (248).
Finalement, la p53 aurait également la possibilité de réguler négativement la transcription de cdc2 via le facteur de transcription NF-Y (249) (Figure 14).
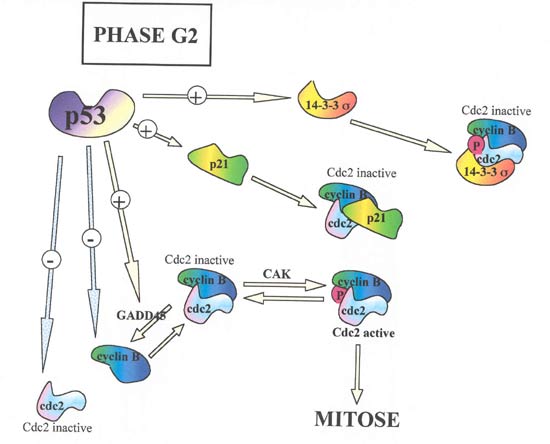
3.3.2.2. La protéine p53 et la réparation de l'ADN :
Après l'arrêt du cycle cellulaire, la cellule ayant subi des altérations au niveau de l'ADN, la protéine p53 active la transcription de gènes (comme GADD45) nécessaires à la réparation des lésions de l'ADN engendrées par le stress (Figure 13). La protéine GADD45 ainsi exprimée, agit par différents mécanismes :
- La protéine GADD45 stimule la réparation par excision de l'ADN via PCNA (250, 251). Elle se lie au PCNA, facteur auxiliaire des ADN polymérases κ et κ, lui-même impliqué dans la réplication, ainsi que dans la réparation de l'ADN (réparation par excision de nucléotides, réparation des mismatch).
- La protéine GADD45 peut aussi inhiber l'entrée en phase S de la cellule (252).
- GADD45 a également la capacité de reconnaître un état altéré de la chromatine et peut aussi moduler l'accessibilité de l'ADN pour les protéines cellulaires (253).
La protéine p53 est également capable de recruter directement les facteurs XPC et TFIIH intervenant dans l'excision des nucléotides de l'ADN endommagé (254). Mais la protéine p53 semble avoir une implication plus directe dans la réparation de l'ADN. En effet, elle possèderait une activité exonucléasique qui pourrait agir comme un correcteur d'erreur (255). Mais cette activité exonucléasique est remise en question dans une plus étude récente (256).
3.3.2.3. La p53 et l'apoptose :
Enfin, si les lésions de l'ADN de la cellule altérée sont trop importantes, la protéine p53 peut induire l'apoptose de la cellule en activant l'expression de gènes pro-apoptotiques comme Bax (257), IGF-BP (258), CD95/Fas (259)...
La protéine p53 semble activer la machinerie apoptotique via le largage du cytochrome c de l'espace inter-membranaire mitochondrial vers le cytoplasme, ce qui déclencherait la cascade des caspases aboutissant à la mort cellulaire. La p53 induirait aussi l'activation directe de certaines caspases (260, 261).
La protéine p53 peut également inhiber la transcription de gènes anti-apoptotiques comme Bcl-2 qui protègent la cellule de l'apoptose (262).
En l'absence de la protéine p53, la cellule aura donc tendance à accumuler plus facilement des mutations pouvant induire une multiplication anarchique des cellules.
3.3.3. Inhibition cellulaire et physiologique de la p53 :
HDM2 (Human Double Minute souvent appelé MDM2 pour Murine Double Minute 2) semble être l'inhibiteur principal de la p53. Cette protéine se fixe sur le domaine de transactivation de la p53. HDM2 inhibe la capacité de la p53 à transactiver la transcription de gènes placés sous sa dépendance (263) par plusieurs mécanismes :
HDM2 se fixe directement sur le domaine de transactivation de la p53, bloquant ainsi l'association de la p53 avec des facteurs associés à la TBP (TATA binding protein), TAFII70 et TAFII31 nécessaires à la transactivation de la transcription (264, 265).
HDM2 peut également induire la dégradation de la protéine p53 via le système du protéasome après ubiquitination (263).
HDM2 aurait également la capacité d'inhiber le processus d'acétylation subi par la p53, normalement effectué par la protéine p300. HDM2 forme un complexe ternaire stable avec p53 et p300 entraînant une inhibition de la fonction de la p53 (266).
La transcription du gène HDM2 est activée par la p53. Le système de dégradation de la p53 induite par HDM2 permettrait le maintien d'une concentration faible de la protéine p53 dans les cellules normales (267), fournissant une boucle d'autorégulation négative (268).
Il existe, en outre, un grand nombre de protéines cellulaires capables d'inhiber de façon physiologique la protéine p53 comme les protéines : phosphatidylinositol 3-OH-Kinase (Akt) (269), Pirh2 (270), HSIR2SIRT1 (pour Silent Information Regulator 2) (271), RB18A (272).
3.3.4. Altérations de la p53 :
L'altération de la fonction de la p53 est un événement très courant dans les cancers. Dans environ 50% des cancers primitifs humains, le gène de la p53 est porteur de mutations (273) en particulier sur les exons 5 à 8, correspondant à sa zone de liaison à l'ADN, altérant par conséquent sa fonction de facteur de transcription. Dans d'autres cas, l'activité de la p53 est inhibée par liaison avec des protéines virales. Par exemple, les oncoprotéines E6 des Papillomavirus humains (HPV) 16 et 18 (274), l'antigène large T du virus simien (SV40) (275), la protéine E1B de 55 kDa des adénovirus (276), la protéine X (HBx) du virus HBV (virus humain de l'Hépatite B) (277), la protéine IE-86 du CMV (Cytomégalovirus) (278), et la protéine BZLF1 du virus d'Epstein Barr (EBV) (279), sont capables de se lier à la p53 humaine inhibant ainsi son contrôle de l'arrêt du cycle cellulaire après dommage de l'ADN (Figure 15). Mais ces protéines virales, se liant toutes à la p53, développent des stratégies très différentes pour l'inhiber.
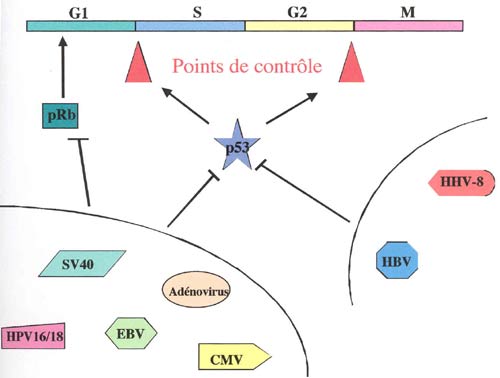
EBV : virus d'Epstein-Barr, CMV : cytomégalovirus, HPV : papillomavirus humains, HBV : virus de l'hépatite B, HHV-8 : herpèsvirus humain de type 8. SV40 : virus simien 40, PRb : protéine du Rétinoblastome.
3.4. Rappels sur NFkB
3.4.1. Généralités :
NFkB est une famille de facteurs de transcription structurellement proches, intervenant dans plusieurs mécanismes physiologiques tels que les réponses inflammatoires, la multiplication cellulaire ou l'apoptose (280). Ils sont formés de plusieurs sous-unités appartenant à la famille Rel/NFkB qui présente 5 membres humains: p50/p105 (ou NFkB1), p52/p100 (ou NFkB2), p65 (Rel A), RelB et c-Rel. Le gène de 8,1 kb codant la protéine p65 est composé de 10 exons. La protéine p65 comprend 551 acides aminés pour un poids moléculaire de 65 kDa. La région N-terminale de p65 contient un domaine d'homologie Rel (DHR) comme toutes les protéines Rel suivie par une séquence de localisation nucléaire (SLN) (Figure 16) qui est essentielle pour le transport du complexe NFkB actif dans le noyau. Cette séquence DHR contient un domaine de fixation à l'ADN (DLA) ainsi qu'un domaine de dimérisation (DD). Cette séquence DHR est, par conséquent, responsable de la dimérisation du facteur, de la localisation nucléaire et de la liaison à l'ADN du complexe NFkB. La région N-terminale de p65 contient un domaine leucine zipper et un domaine de transactivation de la transcription (DTT) nécessaire pour l'activation de la transcription des gènes sous la dépendance de NFkB. NFkB est un facteur de transcription utilisant un motif hélice-boucle-hélice pour lier des éléments de séquences spécifiques dans les régions promotrices des gènes qu'il régule. Un certain nombre de protéines kinases (IKKB, PKA...) peuvent phosphoryler p65 et augmenter ainsi l'activité du complexe NFkB. La phosphorylation du domaine de transactivation de p65 est un événement très important pour le recrutement et/ou l'interaction avec des cofacteurs et des sous-unités de la machinerie transcriptionnelle comme p300 et TFIIB.
(A) Sous-unités humaines appartenant à la classe 1 : p50/p105 ; p52/p100. (B) Sous-unités humaines appartenant à la classe 2 : p65/RelA, RelB et c-rel. Les protéines des deux classes présentent le même domaine d'homologie à Rel (DHR) contenant le domaine de liaison à l'ADN (DLA) ainsi que le domaine de dimérisation (DD). Par contre, seules les protéines de la classe 2 possèdent le domaine de transactivation de la transcription (DTT).
Seules les protéines p65, RelB et c-Rel présentent un domaine de transactivation de la transcription (DTT) potentiel, à l'intérieur de leur séquence C-terminale (281). Ce DTT est riche en sérines ainsi qu'en acides aminés acides et hydrophobes essentiels pour l'activité de transactivation. P50 et p52 ne possèdent pas de DTT et par conséquent ne pourraient pas agir seuls comme facteurs de transcription. Le facteur NFkB actif est un dimère ; le plus souvent constitué de p65 et de p50 ; p50 utile pour la fixation du complexe sur l'ADN et p65 responsable de l'activité transactivatrice de la transcription de NFkB (281). Mais il existe aussi des homodimères p50/p50 et p65/p65 ainsi qu'un certain nombre de combinaisons faisant intervenir chacun des membres de la famille de Rel. L'interaction du complexe rel/NFkB avec l'ADN nécessite la dimérisation des sous-unités NFkB.
Dans la cellule, le facteur de transcription NFkB est séquestré par une protéine appelée IkB (ankyrin rich Inhibitory kappa B protein) (282) qui cache par sa liaison, la séquence de liaison à l'ADN et la séquence de localisation nucléaire de NFkB. Ce complexe NFkB/IkB est localisé dans le cytoplasme (Figure 17). Le processus d'activation de NFkB, induit par des stimuli comme le TNFa (Tumor Necrosis Factor) et l'interleukine 1, est basé sur des phosphorylations d'IkB aboutissant à la dégradation d'IkB entraînant la dissociation du complexe et par conséquent la libération de NFkB (283). Après phosphorylation de deux résidus sérines (32 et 36) dans la partie N-terminale d'IkB par la kinase IKK (IkB Kinase), IkB est polyubiquitynilé et dégradé par le système du protéasome (284-286). NFkB ainsi activé va transloquer à l'intérieur du noyau et va se lier à des séquences d'ADN spécifiques appelées sites kB afin d'activer la transcription des gènes sous la dépendance de NFkB. NFkB se lie à une séquence kB spécifique nucléotidique décamérique ggg ACT TTC C à l'intérieur de la séquence intronique activatrice du gène de la chaîne légère de l'immunoglobuline kappa dans les cellules B immatures, plasmatiques et pré-B (287). Mais il peut se lier à un certain nombre de sites kB contenant une séquence consensus ggg RNN YYC C (R=purine et Y=pyrimidine) à l'intérieur de promoteurs ou de séquences activatrices d'un grand nombre de gènes dans certains types cellulaires. Une fois lié à son motif kB sur l'ADN, le complexe NFkB se lie à des facteurs associés à l'ADN comme ceux appartenant à la machinerie transcriptionnelle (TBP, TFIIB ou CBP/p300). NFkB agirait en synergie avec d'autres facteurs de transcription tels que c-jun ou Sp1. Cela suggère que la régulation différentielle de l'expression de gènes sous la dépendance de NFkB proviendrait d'une combinaison de sites de fixation différents avec des facteurs de transcription différents.
NFkB est capable d'induire la transcription d'un grand nombre de gènes comme des gènes de chimiokines (288, 289), d'interleukines (290-293), du TNF a et b (294), des gènes anti-apoptotiques : bcl-2 (295), bcl-xL (296), des gènes de facteurs de transcription : p53 (297), STAT 5a (171). NFkB est également capable d'induire l'expression de la cycline D1 permettant ainsi l'hyperphosphorylation de la pRb et la progression du cycle de la phase G1 vers la phase S (298, 299).
La voie de signalisation induite par le complexe NFkB est activée par différents stimuli tels des cytokines pro-inflammatoires, des toxines bactériennes (LPS, exotoxine B), des produits de gènes viraux, ou par des stimuli pro-apoptotiques ou nécrotiques (radicaux libres, UV, radiations g).
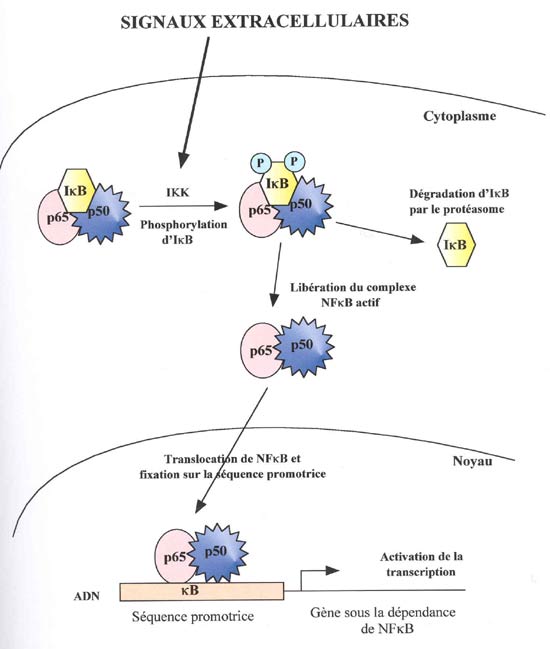
Représentation du mode d'action du complexe NFkB dans la transactivation de la transcription de gènes sous sa dépendance, suite à des signaux extracellulaires.
3.4.2. Altérations de NFkB :
Dans de nombreux cancers primitifs, ainsi que dans un certain nombre de lignées cellulaires tumorales, il existe une activation constitutive de NFkB facilitant la survie cellulaire et la progression tumorale (300-302). En effet, l'activation anti-apoptotique du facteur NFkB est liée au contrôle de l'expression de plusieurs gènes impliqués à différents niveaux de l'apoptose. NFkB induit l'expression d'inhibiteurs de l'apoptose, supprimant l'activation de la cascade des caspases.
L'équipe de Bargou a décrit un niveau élevé d'activité du facteur NFkB dans des cellules dérivées de patients atteints de lymphome de Hodgkin (166). Bargou et al. ont démontré la présence de sous-unités p50 et p65 ainsi qu'une activité constitutive du facteur de transcription hétérodimèrique NFkB p50/p65 dans ces cellules. Ils ont également prouvé que cette activation constitutive de NFκB était nécessaire à la prolifération et à la survie des cellules tumorales de Hodgkin (167). Krappmann et al. ont décrit, dans certains cas du lymphome de Hodgkin, l'existence d'une activité aberrante de l'inhibiteur IkB pouvant expliquer l'activation constitutive de NFkB (168). Cabannes et son équipe ont mis en évidence des mutations sur le gène d'IkB chez 2/8 (25%) des patients atteints de lymphome de Hodgkin pouvant expliquer l'activité aberrante d'IkB (169). De même, Jungnickel et al. ont démontré la présence de mutations somatiques délétères sur le gène d'IkBa dans les cellules de H/RS de 2/4 (50%) des patients atteints de LH indiquant un rôle possible d' IkBa dans la pathogénicité de certains cas de LH (303).
Certains virus sont également capables de transactiver NFkB :
La protéine IE1 du CMV a la capacité d'activer NFkB. Ainsi, Jiang et al. ont montré que la protéine virale IE1 pouvait activer NFkB, mais de façon sélective (304). En effet, IE1 ne pourrait pas induire l'activation de la transcription de Bcl-(x)L, gène cible de NFkB, bien qu'il soit capable de transactiver un gène rapporteur CAT (chloramphenicol acetyl transferase) sous la dépendance de 2 copies d'éléments de réponse à NFkB. Pendant les infections par le CMV, les promoteurs des sous-unités classiques de NFkB (p65-p50) sont transactivés (305). Les protéines virales très précoces IE1-72, IE2-55 et IE2-86 semblent être impliquées dans cette activation. L'infection virale entraîne une augmentation de l'expression du facteur de transcription Sp1 et une augmentation de son activité. Yurochko et al. ont démontré que les protéines IE1-72, IE2-55 et IE2-86 coopèrent avec Sp1 en s'y liant pour augmenter la transactivation des promoteurs de NFkB (306).
La protéine LMP1 (Latent Membrane Protein) de l'EBV peut induire une activité du complexe NFkB dans des lignées de cellules non lymphoïdes et dans des lignées de cellules lymphoïdes B. LMP1 peut également induire, de façon efficace, l'expression du complexe NFkB dans les cellules lymphoïdes T et dans les cellules monocytaires. Les complexes NFkB induits par LMP1 sont capables de se lier à la séquence consensus kB dans le promoteur du gène du récepteur à l'IL2 et peuvent induire l'expression d'un gène sous la dépendance d'un promoteur minimal lié à 4 copies de cette séquence tandem. Cette étude a suggéré un mécanisme possible dans lequel LMP1 pourrait induire l'activation et la prolifération des cellules T (307). Baran-Marszak et son équipe ont comparé les profils d'expression de gènes cellulaires entre des lignées cellulaires infectées par l'EBV et des lignées cellulaires non infectées. Les auteurs ont conclu de cette étude que le facteur de transcription NFkB est une cible potentielle de l'EBV (308).
Rta, protéine virale d'HHV-8, nécessaire au passage d'un état latent à un état réplicatif du virus, aurait la capacité de stimuler une partie seulement des gènes sous la dépendance de différents éléments de réponse à NFkB. La protéine virale Rta ne ferait pas intervenir une forme d'activation classique de NFkB. Mais de façon surprenante, NFkB a également la capacité d'inhiber en partie Rta. La protéine virale Rta, ainsi bloquée par Rel A, ne peut pas transactiver le promoteur du gène de l'IL6 (309).
Travail de Thèse
1.
INTRODUCTION AU TRAVAIL
DE THESE
Notre thématique de recherche de thèse a porté sur l'implication de l'herpèsvirus humain de type 6 (HHV-6) et plus particulièrement, de l'oncogène viral DR7 dans la lymphomogenèse du lymphome de Hodgkin (LH).
Pour cela, nous avons élaboré un travail de recherche que l'on peut diviser globalement en 5 grandes parties.
- Dans un premier temps, nous avons cherché
des séquences d'ADN de l'HHV-6 dans des adénopathies de patients
atteints de lymphomes hodgkiniens et non-hodgkiniens dans le but :
- de démontrer la présence du virus dans ces pathologies,
-
et de mettre en évidence d'éventuelles différences d'incidences
qualitatives ou quantitatives du virus entre ces 2 classes de lymphomes.
Nous avons identifié le type du virus présent et nous avons déterminé la charge virale chez les différents patients atteints de désordres lymphoprolifératifs et porteurs de séquences génomiques d'HHV-6, à l'aide d'une technique de PCR quantitative en temps réel.
- Nous nous sommes intéressés au virus d'Epstein Barr (EBV) décrit comme associé au lymphome de Hodgkin. Nous avons cherché la présence de séquences d'ADN génomique de l'EBV dans les populations précédemment étudiées de patients hodgkiniens (PH) et non hodgkiniens (PNH). Nous avons quantifié le virus dans les adénopathies des patients à l'aide d'une technique de PCR quantitative en temps réel selon la méthode des sondes d'hybridation (Roche).
- Dans un troisième
temps, nous avons séquencé l'oncogène viral DR7
de l'HHV-6, dans les adénopathies de patients atteints de différents
désordres lymphoprolifératifs (lymphomes hodgkiniens et non-hodgkiniens)
et dans des salives d'une population contrôle composée d'adultes
sains. Nous avons ensuite comparé les gènes séquencés
entre les patients et les sujets sains, et enfin entre les patients eux-mêmes,
afin de mettre en évidence d'éventuelles mutations pouvant être
associées aux différents lymphomes.
Nous avons également séquencé les exons 5 à 8 (les plus fréquemment mutés) du gène de la p53 chez les patients atteints de LH, afin de nous assurer de la conservation de ce gène et de sa non-implication dans le développement de cette pathologie. - Au vu des résultats obtenus lors du précédent travail, nous avons considéré que l'implication de l'oncogène DR7 dans la lymphomogenèse du LH, ne faisait pas intervenir des altérations au niveau du gène DR7 (mutations), mais qu'elle pouvait impliquer une dérégulation cellulaire induite par la protéine DR7. L'étude menée par Kashanchi et al. (47) a décrit une liaison entre la protéine DR7 de l'HHV-6A souche U1102 et la protéine p53 humaine aboutissant à l'inactivation de cette dernière. Lors des travaux précédents, nous avons déterminé que le type B de l'HHV-6 était majoritaire dans le LH. Les gènes DR7 de l'HHV-6A et de l'HHV-6B ne présentant entre eux que 42,2% d'homologie (8), il nous a paru intéressant de savoir si DR7B présentait les mêmes potentiels oncogéniques que DR7A. Pour cela, nous avons développé un système double hybride en cellules de mammifères, dans le but de mettre en évidence une éventuelle liaison entre la protéine virale DR7 et la protéine p53 cellulaire. De même, nous avons voulu savoir si la protéine DR7B avait la capacité de fixer et d'inhiber d'autres protéines intervenant dans la régulation du cycle cellulaire comme la protéine du rétinoblastome pRB et la protéine p21WAF1/Cip1.
- La dernière partie de ce travail est basée sur l'étude de l'activation constitutive du facteur de transcription NFκB décrite dans le LH (166, 167). Nous avons voulu savoir si l'HHV-6B pouvait contribuer à cette activation constitutive. Pour cela, nous avons cherché si l'infection par l'HHV-6B pouvait induire une augmentation de la transcription des sous-unités p50 et p65 composant le complexe NFκB, ainsi qu'une sur-activation du complexe NFκB. Et nous avons recherché des transactivateurs viraux capables d'induire l'augmentation de la transcription des gènes cellulaires p50 et p65, et/ou la sur-activation du complexe NFκB.
2. PARTIE 1
2.1. ARTICLE 1 :
Quantification
par PCR en temps réel, technologie TaqMan
et applications en virologie.
Virologie
Novembre-décembre 2001, Vol. 5, n° 6, p439-48.
2.1.1. Introduction :
Dans un premier temps, nous avons fait le point sur les différentes méthodes existantes de PCR quantitative en temps réel. Il nous a paru intéressant de développer nos propres systèmes de quantification avec ces nouvelles techniques. Cet article nous a permis de connaître les différentes chimies et stratégies afin d'en maîtriser au mieux les paramètres. Nous avions le projet de développer plusieurs méthodes de quantification, et nous avons eu l'opportunité de tester une partie des différentes chimies ainsi que différentes stratégies existantes à l'heure actuelle.
Cet article détaille les différentes méthodes de quantification en temps réel et les applications les plus utilisées en virologie.
2.1.2. Discussion :
De plus en plus d'articles sont publiés sur la quantification des génomes viraux depuis le développement des techniques de PCR en temps réel. Ces techniques sont beaucoup plus simples et plus rapides à développer que les techniques de PCR compétitives utilisées jusque-là pour les PCR quantitatives. Globalement, 2 types de chimie sont applicables à la méthode de PCR en temps réel : celle des intercalants et celle des sondes.
- La chimie des intercalants est basée sur l'utilisation de marqueurs fluorescents (le Sybr Green, SG, le plus souvent) qui se fixent dans la double hélice d'ADN en cours de synthèse lors de la PCR. Ce système est souvent utilisé car la mise au point est facile à réaliser et son emploi est peu onéreux. Mais son inconvénient majeur réside dans son manque de spécificité. En effet, le SG se fixe également à des dimères d'amorces ou à des produits synthétisés non spécifiques.
- La chimie des sondes comprend majoritairement 3 techniques différentes : la méthode TaqMan, celle des tests d'hybridation et les sondes beacons. Les deux premiers exemples sont les plus employés actuellement. Ces techniques permettent d'augmenter la spécificité de la réaction du fait de la fixation d'une sonde sur les amplicons, mais elles sont beaucoup plus onéreuses à développer et à utiliser. Pour le TaqMan et les tests d'hybridation, un grand nombre de critères sont à prendre en compte tant pour la détermination de l'amplicon que pour le choix des amorces et des sondes. Les critères à respecter sont moins nombreux pour le TaqMan que pour les tests d'hybridation du fait de la présence d'une seule sonde contre deux pour les tests d'hybridation.
Nous avons développé trois méthodes de quantification différentes au cours de ce travail :
- Nous avons tout d'abord mis au point une technique de quantification du génome de l'HHV-6 avec la technologie TaqMan sur les appareils Applied Biosystems 5700 et 7700 (Courtaboeuf, France). Nous avons appliqué ce système à des populations différentes et sur des échantillons variés. Nous avons choisi en premier le système TaqMan, car il nous a paru plus intéressant de travailler avec une méthode plus spécifique et il nous a semblé, pour un début, que les paramètres à respecter pour l'établissement du système était plus simple.
- Dans un deuxième temps, le laboratoire s'étant équipé d'un appareil LightCycler (Roche, Meylan, France), nous avons donc développé deux systèmes de quantification d'ARNm cellulaires à l'aide du Sybr Green sur LightCycler. Au départ, ces techniques devaient être développées en chimie des sondes, mais lors des premiers tests, qui sont toujours réalisés en Sybr Green, nous avons pu remarquer la grande spécificité des deux systèmes mis en place, à l'aide des courbes de fusion. Nous avons par conséquent conservé le Sybr Green, nous permettant de réaliser un gain de temps et d'argent.
- Enfin, nous avons mis au point un système de quantification du génome de l'EBV avec la chimie des sondes d'hybridation (Roche) sur LightCycler. Nous avions pu acquérir le logiciel « LightCycler Probe Design Software » (Roche) permettant de paramétrer les systèmes de quantification à l'aide des sondes d'hybridation. Après avoir appris à maîtriser ce logiciel, nous avons mis en place un système de quantification du génome de l'EBV que nous avons testé sur les différentes populations de patients sur lesquelles nous avions préalablement travaillé.
Il est intéressant de travailler avec ces méthodes de PCR quantitative en temps réel qui sont très souples et qui ont, par conséquent, un grand nombre d'applications possibles. C'est pourquoi, il existe actuellement beaucoup de techniques de PCR en temps réel, utilisables dans des secteurs très variés et leur développement ne cesse de progresser.
2.2. ARTICLE n° 2 :Real-Time
PCR for quantification of human herpesvirus 6
DNA from lymph nodes and saliva.
Journal of clinical microbiology
July 2002, p2445-2451.
2.2.1. Introduction :
Nous avions à notre disposition un grand nombre d'adénopathies de patients atteints de lymphome de Hodgkin (LH) et de lymphome non hodgkinien. Il s'agit d'un travail rétrospectif, ce qui explique le grand nombre d'échantillons. Les différents prélèvements avaient été réalisés à des fins de diagnostic et avec l'accord des patients (les protocoles de recherche relevant de la loi Huriet). Le travail avait été commencé avec le service d'anatomo-pathologie du CHRU Dupuytren de Limoges, qui s'était occupé de faire les diagnostics des différents lymphomes sur lesquels nous avons travaillé.
L'objectif de ce travail était, dans un premier temps, de savoir en quelle proportion le virus de l'HHV-6 était présent dans les adénopathies de ces différents patients, et de connaître le type viral majoritaire. Enfin, nous nous sommes intéressés à la quantification du génome viral. C'est la raison pour laquelle nous avons développé une technique de PCR quantitative en temps réel, nous permettant de détecter la présence du virus et de déterminer sa charge virale en une seule manipulation. Les prélèvements de ces patients étant très précieux, cela nous a permis de limiter la consommation des extraits d'ADN provenant de ces biopsies.
Il nous a semblé important d'étudier l'HHV-6 dans une population regroupant des désordres lymphoprolifératifs variés, composée de patients atteints de lymphome de Hodgkin, de lymphome B et de lymphome T et NK.
Il est intéressant de détecter la présence du virus dans ces pathologies mais il nous a paru plus important de déterminer la charge virale afin de savoir si éventuellement, le virus pouvait être impliqué dans cette pathologie en tant que virus latent ou réplicatif. De nombreuses études ont permis de détecter la présence du virus dans des adénopathies de patients atteints de divers lymphomes. Mais il existe très peu de travaux portant sur la quantification de l'HHV-6 dans les lymphomes. C'est pourquoi, nous avons mis au point une technique de PCR quantitative en temps réel selon la méthode TaqMan. La détermination de la charge virale nous a permis d'apprécier l'état réplicatif ou latent du virus dans ces pathologies.
Nous avons travaillé sur 2 populations de patients atteints de désordres lymphoprolifératifs et sur une population de sujets sains. Le premier groupe était composé de 37 patients atteints de lymphome de Hodgkin avec une grande majorité de sous-types scléro-nodulaires (83,7%). Le deuxième groupe était composé de 49 patients atteints de lymphomes non hodgkiniens dont 36 sont des lymphomes B et 13 des lymphomes T et NK. La population contrôle était composée de 34 adultes sains. Nous avons quantifié l'HHV-6 dans les adénopathies des deux populations de patients atteints de lymphomes et dans les salives des sujets sains.
L'ADN des biopsies provenant des patients a été extrait utilisant une méthode classique à base de phénol chloroforme. Nous n'avons pas utilisé des systèmes plus simples ou plus rapides comme les kits d'extractions de Qiagen, du fait du caractère précieux de ces échantillons. Nous avons, par la suite, cherché par spectroscopie la présence de phénol et nous avons amplifié un gène cellulaire de ménage afin de détecter la présence d'éventuels inhibiteurs, car les techniques de PCR quantitative, et notamment celles réalisées avec les chimies et l'appareils Roche, sont très sensibles aux inhibiteurs de la polymérisation.
2.2.2. Discussion et conclusion :
Plusieurs résultats se dégagent de ce travail :
- Nous avons établi, dans cette étude, la présence du virus de l'HHV-6 respectivement chez 35,1%, 22,2% et 23,1% des patients atteints de LH, de lymphome B et de lymphome T/NK.
- L'HHV-6 de type B a été retrouvé majoritairement dans 91,7% des cas de tous les patients atteints de lymphomes.
- La distribution de la charge virale était comprise entre 100 à 864.640 copies virales/κg d'ADN pour les lymphomes hodgkiniens et de 100 à 44.290 copies virales/κg d'ADN pour les lymphomes non-hodgkiniens (LNH).
- La distribution de la charge virale est intéressante, mais elle ne représente pas vraiment l'étalement au sein des différentes populations. En effet, parmi les patients atteints de LH, la valeur maximale de 864.640 copies virales/κg d'ADN est une valeur s'écartant considérablement de celles obtenues pour le reste de la population. Il nous a paru judicieux de calculer les médianes afin de prendre en compte la disparité des résultats.

D'après les résultats obtenus dans cette étude, nous pouvons dire que le virus semble plus présent dans les cas de patients atteints de LH. En effet, HHV-6 est présent dans environ 35% des cas de patients atteints de LH contre environ 23% des cas de patients atteints de LNH.
Par contre, il est difficile de déterminer précisément l'état du virus. L'examen de la distribution de la charge virale nous semble en faveur d'un état réplicatif du virus dans les adénopathies de patients atteints de lymphome de Hodgkin, mais également dans le cas des lymphomes T. Il serait intéressant de rechercher la présence d'ARNm viraux dans les adénopathies des patients, de façon à vérifier ces hypothèses, mais les différentes congélations/décongélations des biopsies ont certainement altérées les ARNm se trouvant dans ces prélèvements. Au cours de cette étude nous avons étudié l'effet d'un cycle de congélation/décongélation sur des ADN. Nous avons comparé les résultats de quantification du génome viral dans des extraits d'ADN de salive de sujets sains avant et après un cycle de congélation à -80°C. Nous avons déterminé que ce cycle de congélation entraînait de façon très reproductible une diminution d'un facteur 10 du nombre de copies d'ADN viral dans la quasi-totalité des extraits. Un travail prospectif est, par conséquent, envisagé dans le laboratoire ce qui nous permettrait de travailler sur des prélèvements « frais », donc non altérés.
Nous avons également démontré que, dans nos 3 populations de patients atteints de lymphome et dans la population contrôle, le type B de l'HHV-6 est majoritairement présent. Nous avons détecté le type B dans 100% des sujets sains et dans 91,7% des cas de patients atteints de lymphomes. Le type A a été trouvé chez un patient atteint de LH et chez un patient atteint de LNH B. Ces données sont compatibles avec celles de la littérature. En effet, dans deux études différentes, les taux de détection du type viral B présent au sein d'une population de patients atteints de désordres lymphoprolifératifs étaient respectivement de 92,3% et 94% (78, 222).
Notre population de patients atteints de LH est composée majoritairement du sous-type scléro-nodulaire avec une moyenne d'âge de 39 ans ce qui est compatible avec certaines études montrant que le virus HHV-6 se trouve majoritairement dans le sous-type scléro-nodulaire et chez des patients plutôt jeunes. Di luca et al. ont montré la présence du virus dans 69,2% des cas de LH à sous-type scléro-nodulaire et dans 23% pour les sous-types à cellularité mixte (78). Valente et al. ont détecté des séquences d'ADN génomique viral dans 73,1%, 50% et 70% des sous-types scléro-nodulaire, interfolliculaire et à cellularité mixte respectivement (222). Hallas et al. ont mis en évidence la présence de séquences d'ADN viral dans un pourcentage élevé de patients ayant moins de 60 ans (54%) que chez des patients plus âgés (35%) (75).
Par conséquent, nous avons pu déterminer et établir un certain nombre de constats concernant le virus HHV-6 dans les adénopathies de patients atteints de lymphomes et ce à l'aide d'une seule technique. Globalement, nous pouvons conclure de cette étude :
- L'HHV-6 se présente avec une fréquence plus importante chez les patients atteints de LH
- Le type viral associé à cette pathologie est très majoritairement le type B.
- Le virus HHV-6 se trouve dans un état plutôt réplicatif dans les adénopathies de patients atteints de lymphomes de Hodgkin associés à l'HHV-6B.
- L'HHV-6B est détecté plus particulièrement dans les sous-types scléro-nodulaires du LH.
- L'HHV-6 se trouve préférentiellement chez des patients atteints de LH relativement jeunes.
3. PARTIE 2
3.1. Détection du virus d'Epstein-Barr
3.1.1. Introduction :
Dans la dernière partie de ce travail de thèse, nous avons recherché la présence du virus d'Epstein-Barr (EBV) dans les adénopathies des patients atteints de différents désordres lymphoprolifératifs (patients sur lesquels nous avons travaillé dans les études précédentes). Il nous a paru important de rechercher l'EBV décrit comme associé au LH, de façon à ne négliger aucun paramètre dans notre étude de l'implication de l'HHV-6 dans le lymphome de Hodgkin.
Nous avons développé une technique de PCR quantitative en temps réel sur LightCycler avec la chimie des sondes d'hybridation (Roche), dans le but de quantifier le virus EBV dans les adénopathies des patients.
Le test d'hybridation nécessite l'utilisation de deux sondes : l'une donneuse d'énergie, A, et l'autre accepteuse, B, conçues pour reconnaître deux séquences adjacentes sur le fragment d'ADN amplifié. L'éloignement spatial de ces deux sondes ne permet pas de transfert d'énergie de A vers B, et seule A émet une fluorescence après excitation. Au cours de l'amplification, les 2 sondes s'hybrident sur l'amplicon permettant un transfert d'énergie de A vers B entraînant, après excitation une émission de B seule. La fluorescence de B est mesurée en fin de phase d'hybridation de la PCR. La quantité de fluorescence émise par B est, par conséquent, directement proportionnelle à la quantité d'amplicons synthétisés.
3.1.2. Matériel et méthodes
Nous avons travaillé sur les adénopathies de 35 patients atteints de LH, 36 patients atteints de lymphome B et 11 atteints de lymphome T et NK (310). La moyenne d'âge est respectivement de 43,4 ans, 64,7 ans et 63,8 ans et le ratio homme/femme respectivement de 19/18, 16/20 et 11/2 pour les patients atteints de LH, de LB et de LT.
Les extraits d'ADN avaient été obtenus à l'aide d'une technique classique d'extraction au phénol chloroforme (310). Les amorces (EB1 et EB2) (Qbiogene, Strasbourg, France) et les sondes (EB3 et EB4) (Proligo, Paris, France) rapportées dans le tableau 15 ont été choisies sur le gène de la protéine majeure de capside (MCP) de l'EBV à l'aide du logiciel « LightCycler Probe Design Software » (Roche, Meylan, France). Nous avons travaillé sur 500 ng d'ADN de biopsies. Le volume réactionnel final de 20 κL comprend 0,9 κM de chacune des deux amorces EB1 et EB2, 4 mM de MgCl2, 200 nM de la sonde marquée à la fluorescéine (EB3), 400 nM de la sonde marquée au Red 640 (EB4), 1X de mix (FastStart DNA Hybridization Mix, Roche). Les cycles réalisés pour l'amplification sont répertoriés dans le tableau 17.
Le fragment de 100 pb du gène viral MCP a été cloné dans le vecteur PCR2.1 à l'aide du kit TOPO Kit (Invitrogen, Cergy Pontoise, France). Les plasmides ont été extraits à l'aide du QIAprep Miniprep kit (Qiagen, Courtaboeuf, France). La quantité d'ADN plasmidique a été évaluée par spectrophotométrie à 260 nm et la pureté des extraits par le rapport Absorbance à 260 nm/ Absorbance à 280 nm devant être compris entre 1,7 et 1,9.
Une gamme d'ADN plasmidique a été réalisée allant de 107 à 10 copies/5 κl dans un volume final de 500 κL d'eau distillée. Cette gamme a été conservée à 4°C, afin d'éviter la destruction de l'ADN due à des étapes de congélation-décongélation successives.
3.1.3. Résultats :
Des tests de spécificité ont été réalisés avec des ADN provenant des virus HHV-6, HHV-7, CMV, HHV-8, HSV-1, HSV-2 et VZV. Aucune accroche non spécifique n'a été détectée. Des tests de reproductibilité intra- et inter-essais ont également été réalisés avec de bonnes corrélations. Les résultats de quantification pour tous les patients sont répertoriés dans la figure 18 et dans le tableau 16.
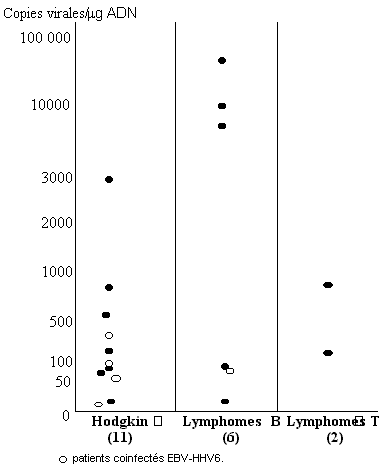
Nous avons quantifié des séquences d'ADN de l'EBV chez 11/35 (31,4%) des patients atteints de LH, chez 6/36 (16,7%) des patients atteints de lymphome B et chez 2/11 (18,2%) des patients atteints de lymphome T et NK.

3.1.4. Discussion/ Conclusion :
Les pourcentages de détection obtenus à l'aide de cette technique sont assez faibles par rapport à ceux décrits dans d'autres auteurs. En effet, les séquences d'EBV sont trouvées chez des patients atteints de LH dans 40 à 50% des cas (185). D'après notre étude, seulement 31,4% des patients atteints de LH présentent des séquences de l'EBV. Les biopsies sur lesquelles nous travaillons sont des produits anciens et qui ont été congelées plusieurs fois pouvant expliquer que l'ADN soit peu altéré. Néanmoins, ces résultats pourraient également s'expliquer par le fait que 86,1% de nos patients atteints de lymphome de Hodgkin sont de sous-types scléro-nodulaires. En effet, Shibata et al. a démontré que l'EBV était plus souvent détecté dans le sous-type à cellularité mixte (80%) (189).
Néanmoins, Jarrett et al. ont montré que la plupart des patients atteints de lymphome de Hodgkin et porteurs de séquences de l'EBV sont, soit des enfants, soit des adultes de plus de 50 ans (199). Dans notre étude, la moyenne d'âge de la totalité des patients atteints de lymphome de Hodgkin est environ de 39 ans ; celle des patients atteints de LH et porteurs de séquences d'ADN du virus EBV est de 56 ans environ.
Si nous comparons les données obtenues lors de la détection du génome de l'HHV-6 chez les patients atteints de LH avec ceux de la détection du génome de l'EBV dans cette même population, nous nous apercevons que seulement 4/11 (36,3%) des patients sont co-infectés par les virus HHV-6 et EBV. Pour les lymphomes B, seulement 1/6 (16,7%) des patients sont porteurs de séquences d'ADN de l'HHV-6 et de l'EBV. Par contre, aucun patient atteint de lymphome T et NK n'est co-infecté.
Au sein de la population de tous les patients atteints de lymphome, nous remarquons un très faible pourcentage de co-infection, seulement 6/82 (7,3%) des patients sont co-infectés par l'HHV-6 et par l'EBV dont 4/35 (11,4%) des patients avec un LH, et 1/47 (1,8%) des patients avec un LNH. Ces résultats sont très faibles pour ces deux populations de patients atteints de désordres lymphoprolifératifs.
Nous pouvons donc conclure, que dans la population de patients atteints de LH et porteurs de séquences d'ADN génomique de l'HHV-6, seulement une très faible proportion se trouve être co-infectée avec le virus EBV.
4. PARTIE 3
4.1.
ARTICLE n° 3 :
High conservation of HHV-6B DR7 gene and human p53
gene; suspected implication of DR7B onco-protein in the development of Hodgkin's
lymphoma.
Soumis à Journal of Medical Virology
4.1.1. Introduction :
Cet article rassemble trois études différentes menées consécutivement.
Nous avons, dans un premier temps, tenté de comprendre comment le virus pourrait être impliqué dans la lymphomogenèse de cette pathologie. Au cours de l'étude précédente, nous avons établi que le virus HHV-6B était fréquemment associé à certains cas de lymphome de Hodgkin, et que ce virus se trouvait en quantité relativement importante dans les adénopathies des patients. Les équipes de Thompson et de Kashanchi ont détecté un oncogène viral appelé DR7, capable de transformer différentes lignées cellulaires et d'induire une tumeur par injection de la protéine chez la souris nude (47, 230). Le gène DR7 nous a paru être un bon candidat pour débuter l'étude du pouvoir oncogène du virus HHV-6B. Nous avons voulu savoir si ce gène viral présentait des mutations, par rapport à la souche virale en culture, qui pourraient être spécifiquement présentes dans les cas de LH. Nous avons, par conséquent, séquencé le gène viral DR7 chez tous les patients infectés et atteints de LH, de LB, de LT mais aussi chez des sujets sains, ainsi que le gène DR7 des souches Z29 (type B) et U1102 (type A) en culture. Nous avons développé un système de nested PCR pour amplifier le gène DR7 du type B. Nous avons utilisé pour les différentes PCR le système High Fidelity Expand System (Roche, Meylan, France) du fait de la grande fidélité de ce mélange d'enzymes. Nous avons séquencé le cadre de lecture complet de DR7 dans les adénopathies de 15 patients atteints de LH, de 9 patients atteints de lymphome B, de 3 patients atteints de lymphome T ainsi que dans la salive de 14 sujets sains à l'aide du séquenceur ABI PRISM 310. Nous avons comparé les différentes séquences obtenues aux séquences du gène DR7B de la souche Z29 déposées dans les banques de données (GENBANK n° NC 000898) (7). Nous avons également séquencé et comparé les gènes DR7 de 2 souches d'HHV-6 en culture : Z29 et U1102.
Dans un deuxième temps, nous avons voulu étudier le degré de conservation du gène de la p53 chez les patients atteints de LH, afin de prendre en compte tous les paramètres pouvant influencer la transformation cellulaire. De rares mutations (11,5%) ont été décrites par certains auteurs sur la zone de fixation à l'ADN du gène de la p53 (125). Nous avons, par conséquent, séquencé les exons 5 à 8 (correspondant à la zone de liaison à l'ADN) du gène de la p53 dans les adénopathies de tous les patients atteints de LH, à l'aide du séquenceur VISIBLE GENETICS (Evry, France).
Dans un troisième temps, nous avons travaillé sur l'aspect protéique de l'oncogène viral DR7. En effet, cette protéine entraîne le développement de tumeurs chez la souris lors qu'elle est injectée. Il est fort probable que le caractère oncogénique de DR7 soit porté par sa protéine. Kashanchi et al., ayant démontré une inhibition de la protéine p53 cellulaire par la protéine DR7 de l'HHV-6 type A (U1102) (47), et les gènes DR7 des types A et B ne présentant que 42,2% d'homologie (8), il nous a paru capital de vérifier si la protéine DR7 du type B avait, elle aussi, la capacité de se fixer à la p53. Nous avons pu voir précédemment que l'inhibition de la protéine p53 était très souvent associée à l'établissement de cancers. Ne disposant pas d'anticorps spécifique de la protéine DR7, nous avons exploré un système double hybride. Nous avons travaillé en cellules humaines de façon à être dans les conditions les plus proches de la réalité. En cellules de mammifères, la protéine p53 peut subir des glycosylations post-traductionnelles et a, par conséquent, l'aspect de la p53 cellulaire produite normalement par la cellule. Nous ne savons pas si la protéine virale DR7 subit des glycosylations, mais elle présente des sites potentiels dans sa structure. Nous avons travaillé avec le système ChechkmateTM mammalian two-hybrid system de Promega (Charbonnières, France). L'ADN complémentaire de la p53 a été fusionné avec le domaine de liaison à l'ADN de GAL4 et le gène viral DR7 avec le domaine de transactivation VP16 (Figure 19). Si les protéines DR7 et p53 se lient l'une à l'autre, cela entraîne un rapprochement du domaine de liaison à l'ADN et du domaine de transactivation reformant ainsi un facteur de transcription actif pouvant agir sur les sites de fixation à GAL4 en amont d'un gène rapporteur de la luciférase (Figure 20). Nous avons révélé l'expression du gène de la luciférase à l'aide du kit Dual-luciferase® reporter assay system. Nous avons mis au point des tests de transfection sur les cellules humaines adhérentes HEp2 à l'aide de l'agent transfectant FuGENE 6 (Roche).
Nous avons également recherché des interactions entre la protéine virale DR7 et des protéines cellulaires, intervenant dans le contrôle du cycle cellulaire comme la protéine du rétinoblastome pRb et la protéine p21Waf1/Cip1.
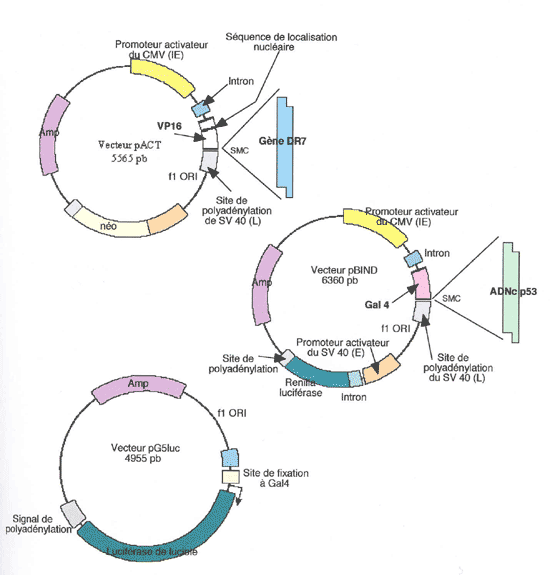
Le gène viral DR7 a été fusionné au domaine de transactivation du facteur VP16 dans le plasmide pACT (Promega), l'ADNc de la p53 au domaine de liaison du facteur GAL4 dans le plasmide pBIND (Promega). Le gène rapporteur de la luciférase est porté par le plasmide pGL5luc (Promega).
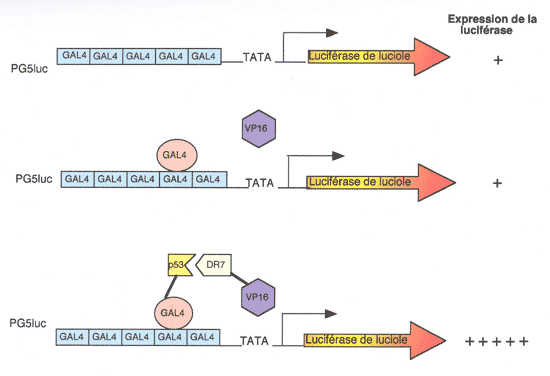
Si les protéines DR7 et p53 se lient l'une à l'autre, cela entraîne un rapprochement du domaine de liaison à l'ADN et du domaine de transactivation reformant ainsi un facteur de transcription actif pouvant agir sur les sites de fixation à GAL4 en amont d'un gène rapporteur de la luciférase sur le plasmide pG5luc (Promega).
4.1.2. Discussion :
Nous avons entièrement séquencé les gènes viraux DR7 des souches de l'HHV-6 retrouvées dans les adénopathies des patients atteints de LH, de lymphome B, de lymphome T/NK ainsi que dans les extraits de salives des sujets sains. Nous avons pu mettre en évidence une grande homologie de séquences des gènes DR7 détectés chez les patients et les sujets sains. Des mutations ont, certes été trouvées, mais aucune n'est associée spécifiquement à un groupe de pathologies. Nous avons globalement déterminé la présence de 2 mutations (I150T) et (V171L) présentes chez presque tous les patients ou sujets sains, induisant un changement d'acide aminé dans la séquence protéique prédictive. De plus, nous avons trouvé trois mutations (V167M), (G152E) (G158R) chez trois patients différents tous atteints de LH ; la mutation (G158R) étant également présente chez un sujet sain. Il est assez intéressant de noter que les personnes porteuses de la mutation (G158R) ne possèdent pas la mutation (I150T) pourtant présente chez toutes les autres personnes testées.
Nous avons donc démontré une grande stabilité du gène DR7 chez les patients atteints de désordres lymphoprolifératifs.
Nous avons également comparé les séquences protéiques prédictives de DR7 pour les souches Z29 et U1102, nous permettant de vérifier la faible homologie entre ces deux protéines, décrite par Isegawa et al. (8). La protéine DR7A U1102, produite par un site d'initiation différent, possède 118 acides aminés de plus que la protéine DR7B Z29. En comparant les séquences communes de ces deux gènes, 34 mutations (transitions et transversions) efficaces, induisant le remplacement d'un acide aminé par un autre, ont été trouvées, ainsi que 9 insertions induisant l'ajout de 3 acides aminés dans la séquence protéique de DR7B Z29. L'insertion de ces 9 acides nucléiques n'a pas engendré de changement de cadre de lecture de la protéine DR7B Z29.
Dans cette partie du travail, nous avons voulu nous assurer de la non-implication de mutations du gène de la p53 dans la lymphomogenèse du LH. Nous avons séquencé les exons 5 à 8 (les plus fréquemment mutés) de 26 patients atteints de LH à l'aide du système de séquençage VISIBLE GENETICS.
Aucune mutation spécifique associée au LH ne fut mise en évidence lors de ce travail. Seules quelques mutations ont été trouvées mais en dehors des exons de la p53. Nous avons pu observer une très grande conservation de ces quatre exons chez les patients atteints de LH, ce qui vient confirmer les travaux d'autres équipes (127, 128).
Le gène de la p53 est muté dans près de 50% des cancers primaires aboutissant à des produits du gène de la p53 le plus souvent inactifs. Les mutations du gène de la p53 constituant un phénomène majeur dans le processus de transformation cellulaire, il était important que nous vérifiions la conservation du gène de la p53 dans les adénopathies des patients sur lesquels nous travaillons. Dans le cas des LH, nous pouvons donc conclure que, si l'inactivation de la p53 intervient dans le LH, cela ne provient pas de mutations présentes sur sa séquence génomique.
Dans cette dernière partie, nous avons voulu savoir si la protéine DR7B, dont le gène ne présente que 42,2% d'homologie avec celui de DR7A (souche U1102) (8), possède les mêmes propriétés oncogéniques que DR7A c'est-à-dire une capacité à se lier à la protéine p53 cellulaire.
Nous avons par conséquent développé un système de double hybride en cellules de mammifères afin de rechercher, d'une part une interaction entre la protéine DR7B et la protéine p53, et d'autre part, entre la protéine virale DR7B et d'autres protéines cellulaires impliquées dans la régulation du cycle cellulaire (pRb et p21).
Nous avons démontré l'existence d'une liaison entre la protéine p53 humaine et la protéine virale DR7B. Les protéines DR7 A et B ont la capacité de se lier à la protéine p53 malgré les faibles homologies présentes sur leur gène respectif. Par conséquent, nous pouvons supposer que la p53 se fixe sur une zone commune de ces deux protéines. DR7A se fixe sur la zone de liaison à l'ADN de la p53 entraînant une inhibition de ses fonctions. Mais les zones communes de ces deux protéines présentent tout de même des mutations, un test d'inhibition des propriétés de la protéine p53 en fonction des liaisons avec les différentes protéines virales est envisagée. En effet, l'affinité de liaison entre ces protéines peut être affectée par la présence de ces mutations.
Par contre, nous n'avons pas observé de liaisons entre la protéine virale DR7B et la p21Waf1/Cip1 ou la protéine du Rétinoblastome (pRb). Les virus oncogènes à ADN ont pour la plupart des protéines capables de lier à la fois la p53 et la pRb. Mais dans d'autres cas, ces propriétés sont portées par des protéines virales différentes. Dans le cas de l'HHV-6, la protéine DR7 n'a pas la capacité de se lier à ces protéines cellulaires. D'autres protéines virales ont certainement ses propriétés. Des tests de co-immunoprécipitations peuvent être envisagés afin de rechercher ces différents partenaires viraux.
4.1.3. Conclusion :
Il ressort un certain nombre de points de ce travail:
- Nous avons démontré l'absence de mutations sur le gène de la p53 retrouvé dans les adénopathies de patients atteints de LH.
- Nous avons également observé l'absence de mutations spécifiques sur le gène viral DR7 pouvant être associées au LH.
- Nous avons montré que l'oncogène viral DR7 était susceptible d'être un bon candidat dans l'établissement du processus de transformation cellulaire du LH. Dans l'étude précédente, nous avions observé, que l'HHV-6 majoritairement présent dans le LH était le type B, et que ce virus semblait plutôt réplicatif dans les adénopathies des patients. La conséquence de ces deux éléments est que la transcription des gènes viraux a probablement lieu induisant la synthèse de la protéine DR7 du type B dans les adénopathies de ces patients.
- Nous avons mis en évidence le fait que la protéine virale DR7B avait la capacité de se lier à la protéine p53, mais pas aux protéines pRb et p21Waf1/Cip1.
Au total, nous nous pouvons donc émettre l'hypothèse suivante, si DR7 est impliqué dans le développement du lymphome de Hodgkin, cela ne provient pas de son gène (du fait de mutations), mais du caractère oncogénique de sa protéine (liaison DR7B/p53).
De ce fait, la suite logique du travail vise à étudier le pouvoir transformant de la protéine DR7B et plus largement de l'HHV-6B. Outre un rôle dans l'inhibition de la protéine p53, il est vraisemblable que la protéine virale transactivatrice DR7B exerce d'autres fonctions intervenant dans la dérégulation du cycle cellulaire.
5. PARTIE 4
5.1. ARTICLE n°
4 :
Human tumor suppressor p53
and DNA virus
Soumis à Reviews in Medical virology
5.1.1. Introduction :
Dans le travail précédent, nous avons établi que la protéine virale DR7B avait la capacité de se lier à la protéine suppresseur de tumeur p53, comme son homologue DR7 du type A, et comme de nombreuses protéines codées par d'autres virus à ADN, liaison ayant pour effet l'inhibition de la p53. Nous avons, par conséquent, voulu connaître les conséquences exactes de l'inhibition de cette protéine. C'est la raison pour laquelle nous avons écrit cette revue sur la p53 et les différentes inhibitions qu'elle subit par les virus à ADN.
Dans cet article, nous avons passé à revue, dans un premier temps, les connaissances actuelles sur la p53 avec ses modes d'actions, ses différents rôles, ses activations et ses inhibitions, et dans un deuxième temps, nous avons étudié les différents virus à ADN ayant une action oncogénique par l'intermédiaire de la p53 avec les différentes stratégies mises en oeuvre par ces virus pour l'inhiber.
Nous avons pu nous rendre compte de la complexité des différentes fonctions de la protéine p53 dans le cycle cellulaire. Elle entraîne l'arrêt du cycle cellulaire du fait de dommages subis par l'ADN, agit sur la réparation de l'ADN et sur l'apoptose. Il s'agit de procédés très complexes sous la dépendance d'une seule protéine. Globalement, son rôle principal est celui de facteur de transcription, mais elle pourrait également agir directement de façon différente sur d'autres processus comme celui de la réparation de l'ADN avec son activité présumée 3'-5' exonucléasique.
Il semble qu'il existe très peu de mécanismes impliqués dans la régulation du cycle cellulaire ne faisant pas intervenir la p53. Environ 50% des cancers primitifs présentent des mutations sur le gène de la p53. Et dans d'autres cas, des protéines virales induisent le même effet que les mutations c'est-à-dire l'inhibition de fonction de la p53. Mais les processus développés par les virus sont différents ; il existe des inactivations de la p53 par interaction directe entre protéine virale et p53, pouvant bloquer la liaison de cette dernière avec l'ADN, ou rendant son domaine de transactivation inactif, voir cacher les séquences de tétramérisation, de phosphorylation ou d'acétylation nécessaires à son activation. Mais l'inhibition de cette protéine peut être plus indirecte avec une sur-activation de son inhibiteur principal HDM2, ou en inhibant les protéines responsables des modifications post-traductionnelles de la p53 (kinases, p300...). Il existe également une inactivation en aval de la p53 : certaines protéines virales pouvant inhiber les protéines dont l'activation est nécessaire à l'action de la p53 et qui collaborent avec la p53 pour l'arrêt du cycle cellulaire.
5.1.2. Discussion / conclusion :
La protéine suppresseur de tumeur p53 interagit avec un grand nombre de protéines codées par des virus à ADN, aboutissant à l'inhibition de ses fonctions dans la régulation du cycle cellulaire. Mais les mécanismes employés par ces protéines virales peuvent être très différents.
La protéine E1B55K et la protéine E4orf6 de l'Adénovirus forment des complexes stables avec la protéine p53 (311, 312) et inhibent ainsi son activité de transactivateur de la transcription. Ces 2 protéines pourraient se lier séparément à la p53, mais elles seraient capables de promouvoir une dégradation rapide de la p53 en se liant toutes les deux à la fois (313).
L'antigène large T du SV40 peut également se fixer sur la protéine p53 inhibant son activité transactivatrice (314, 315).
La protéine E6 des Papillomavirus humains (HPV) de type 16 ou 18 a la capacité de se lier à la p53 et d'inhiber son activité de transactivateur (316). Cette protéine, par sa liaison à la p53, a également la capacité d'induire la dégradation de la p53 (317). Ces deux activités de la protéine virale E6, inhibition de la transcription et dégradation de la p53, seraient complètement indépendantes (316).
La protéine HBx de l'HBV a la capacité de se lier à l'extrémité carboxy-terminale de la p53, entraînant une inhibition des fonctions de cette dernière (318, 319). Il a également été démontré que la protéine core HBc de l'HBV agit comme un répresseur du promoteur humain de la p53. En fait, HBc et HBx agiraient en synergie pour réprimer l'expression de la p53 dans les cellules HepG2 (320).
La protéine EI-86 du CMV peut, par sa liaison à la p53, réduire une partie de l'activité de transactivation de la transcription de la p53 (278). Mais cette inhibition ne semble pas avoir de conséquences sur le contrôle cellulaire au niveau de la transition G1/S (321). Bien que le CMV soit un agent pathogène important causant un certain nombre d'infections primaires et récurrentes très répandues, il n'a encore été relié à aucun des cancers humains répertoriés.
La protéine BZLF1 de l'EBV peut aussi se lier à l'extrémité carboxy-terminale de la p53 (279), entraînant des effets négatifs sur les modifications post-traductionnelles nécessaires à l'activation de la p53.
Ces protéines virales sont également souvent capables de se lier et d'inhiber différentes protéines cellulaires impliquées dans le contrôle du cycle cellulaire telles que la p21WAF1/Cip1 ou la protéine du rétinoblastome (pRb). Par exemple, la protéine virale E7 de HPV16 a la capacité de se lier à la pRb (322) ainsi qu'à la p21 (323) ; de même, l'antigène large T du SV40 peut former un complexe spécifique avec la pRb (324).
Les virus cités dans cet article inhibent tous la p53, mais ne déclenchent pas nécessairement la prolifération anarchique des cellules. Il est probable que des homologues de fonction comme p63 et/ou p73 compensent la perte d'activité de la p53. D'autre part, la cancérisation est un processus mutlifactoriel, n'impliquant pas seulement la p53. De ce fait, si les virus ne déclenchent pas systématiquement la transformation cellulaire, ils y participent de manière plus ou moins active.
6. PARTIE 5
6.1.
ARTICLE n° 5
NFκB constitutive activation
is induced by HHV-6B and
more specifically by DR7 and U3 viral oncoproteins.
Soumis à Virus research
6.1.1. Introduction :
Dans cette partie de la thèse, nous avons travaillé sur le pouvoir oncogène de l'HHV-6B. Dans l'étude précédente, nous avions déterminé que la protéine virale DR7 présentait un potentiel oncogène. Nous avons, par conséquent travaillé sur la protéine DR7. Nous avons également pensé, suite à l'étude réalisée par Mori et al. (46) portant sur la protéine virale transactivatrice U3, que cette protéine pouvait, elle aussi, présenter un certain pouvoir oncogène lié à sa fonction de transactivation et pouvait être un bon candidat pour l'établissement du processus transformant.
Pour cette étude, nous nous sommes intéressée au complexe protéique NFκB dans le lymphome de Hodgkin associé à l'HHV-6. Bargou et al. ont démontré, dans deux études différentes, qu'il existait une activation constitutive de ce facteur de transcription dans des lignées de cellules de H/RS provenant de patients atteints de LH ainsi que dans les cellules de H/RS provenant de fluides péricardiques d'un patient atteint de LH (166). Ils ont aussi observé que cette activité constitutive du facteur de transcription NFκB était nécessaire à la prolifération et à la survie des cellules tumorales de Hodgkin, les protégeant de l'apoptose induite par le stress (167). Nous avons voulu savoir si l'infection par l'HHV-6B pouvait entraîner, dans un premier temps, une augmentation de la transcription des sous-unités p50 et p65 constituant du complexe NFκB, et dans un deuxième temps, une sur-activation de ce même complexe.
Enfin, nous avons cherché si des protéines transactivatrices virales comme DR7 et U3 pouvaient être responsables de ces deux phénomènes. Pour cela, nous avons mis au point différentes techniques de transfection cellulaire sur des cellules humaines adhérentes (HEp 2) et en suspension (MT4), dans lesquelles l'activité de gènes rapporteurs a été mesurée.
Les essais de transactivation de la transcription des sous-unités p50 et p65 ont été réalisés à l'aide de deux types plasmides :
le plasmide pGL3 Enhancer Vector (Promega, Courtabeouf, France) dans lequel nous avons inséré les zones promotrices des gènes humains p50 et p65 (GENBANK n° AH004203 et L01459 respectivement) en amont d'un gène rapporteur de luciférase,
le plasmide pCi (Promega) dans lequel nous avons inséré les ADNc des gènes viraux DR7 et U3.
Les tests de transactivation du complexe NFκB ont été réalisés à l'aide du plasmide pNFκB-Luc (Clontech) contenant des éléments de réponse à NFκB, les séquences κB, en amont d'un promoteur minimal et d'un gène rapporteur de luciférase.
Finalement, nous avons réalisé une technique de transcription inverse (RT), suivie d'une PCR quantitative en temps réel utilisant le Sybr Green (Roche) sur LightCycler, nous permettant de quantifier les ARN messagers des deux sous-unités p50 et p65 du complexe NFκB suite à l'infection virale.
6.1.2. Discussion :
Lors de ce travail, nous avons déterminé que l'infection des cellules MT4 par le virus HHV-6B souche HST entraînait une augmentation de la transcription des sous-unités p50 et p65 du complexe NFκB. Cette augmentation est relativement faible, et d'interprétation délicate. En fait, les extractions d'ARNm des cellules MT4 infectées par l'HHV-6B ont été contrariées par la lyse spontanée des cellules environ 20h après l'infection alors que les cellules non infectées continuaient à se multiplier. Nous avons obtenu une augmentation de la transcription de p50 et p65 après 8 heures d'infection, se prolongeant jusqu'à 20 heures, mais nous n'avons pas pu vérifier celle-ci après 20 heures d'infection car les cellules infectées mourraient, entraînant des rendements d'extraction d'ARNm très mauvais. Nous avons également montré que les protéines virales transactivatrices DR7 et U3 induisaient dans les cellules humaines HEp 2, une augmentation respective de 58,2% et de 164% de la transcription du gène p65 et de 44% et 29,6% de la transcription du gène p50. DR7 et U3 pourraient être responsables des augmentations de p50 et p65 observées dans les cellules MT4 infectées. Il faut également noter que l'augmentation des transcriptions de p50 et p65 débute à partir de 8 heures post-infection, or les protéines virales DR7 et U3 ne semblent être détectées qu'à partir de 18 heures post-infection (46, 47). Par conséquent, d'autres protéines virales pourraient être impliquées dans cette augmentation. Et les protéines DR7 et U3 pourraient prendre le relais à partir de 18 heures.
De plus, nous avons montré que l'infection par l'HHV-6B souche HST entraînait une augmentation de l'activité du complexe nucléaire NFκB, réalisée en partie par les protéines virales DR7 et U3. Ces protéines sont, par conséquent, capables d'augmenter la transcription de p50 et de p65, induisant la formation d'un complexe NFκB actif. L'activité de NFκB semble augmenter de 131% suite à l'expression de la protéine virale DR7 et de 136% avec celle de U3.
L'augmentation des transcriptions de p50 et p65 n'est peut-être pas le seul mécanisme employé par ces protéines pour induire une suractivation du complexe NFκB. Les protéines virales DR7 et U3 pourraient être capables d'augmenter en plus, la dimérisation ou la localisation nucléaire du complexe NFκB. De plus, DR7 et U3 pourraient entraîner une augmentation de l'activité d'autres facteurs cellulaires de transcription ayant la capacité de se lier aux éléments de réponse à NFκB : les séquences κB. Le facteur de transcription NF-A, par exemple, ainsi que d'autres protéines cellulaires ont la capacité de se lier aux séquences κB induisant la transcription du gène en aval (287, 325).
Par conséquent, des études supplémentaires doivent être envisagées, afin d'élucider les rôles complets des protéines virales DR7 et U3.
Les stratégies utilisées par les protéines DR7 et U3 pour activer le facteur de transcription NFκB ne sont pas élucidées. Ces deux protéines sont considérées comme des transactivateurs viraux. Mais agissent-elles directement sur l'élément de réponse κB comme des facteurs de transcription ou agissent-elles comme des recruteurs de facteurs de transcription ? D'après l'étude théorique de leurs séquences protéiques prédictives, elles ne possèderaient pas les séquences spécifiques des facteurs de transcription comme les domaines en doigt de zinc. Des études doivent, par conséquent, être réalisées afin de vérifier si ces protéines virales ont la capacité de se fixer directement sur les séquences κB ou si elles recrutent des facteurs cellulaires de transcription et agissent plus indirectement.
D'autres herpèsvirus ont également la capacité de transactiver le complexe NFκB, comme le CMV ou l'EBV. Dans le cas du CMV, les auteurs montrent que la protéine virale IE1 pourrait activer le facteur de transcription NFκB, mais seulement de manière sélective. Ils ont démontré que cette protéine avait la capacité de transactiver un gène rapporteur sous la dépendance de l'élément de réponse de NFκB, mais elle ne pourrait pas entraîner une augmentation in vivo de la transcription du gène Bcl-(x)L, gène cible de NFκB. Il faut, par conséquent, être prudent et vérifier in vivo la transactivation de gènes cellulaires sous la dépendance de NFκB. De plus, lors de cette étude, les auteurs ont décrit une augmentation de l'activité du facteur cellulaire de transcription Sp1 suite à l'infection par le CMV. Ils ont montré que les protéines virales IE1-72, IE2-55 et IE2-86 coopéraient avec Sp1 pour augmenter la transactivation des promoteurs de NFκB. Il est intéressant de noter que, dans une étude réalisée en 1994, Wang et al. ont déterminé que le fragment ZVH14 de l'HHV-6 avait la capacité de transactiver les LTR du VIH à l'aide du facteur de transcription Sp1 (326). Il est possible que des protéines virales de l'HHV-6 puissent recruter le facteur Sp1 entraînant, comme pour le CMV, une augmentation la transactivation des promoteurs cibles de NFκB. Pour cela, d'autres manipulations sont envisagées afin de déterminer l'implication du facteur cellulaire Sp1 dans le processus de transactivation de NFκB via l'HHV-6B.
Nous pouvons conclure de cette étude que:
- L'infection par l'HHV-6B entraîne une augmentation de la transcription des sous-unités p50 et p65 ainsi qu'une augmentation de l'activité de NFκB.
- Les protéines virales DR7 et U3, décrites comme transactivatrices, ont la capacité de transactiver l'expression de certains gènes cellulaires. Nous avons observé qu'elles agissaient sur les séquences promotrices des sous-unités p50 et p65 mais aussi sur les éléments de réponse à NFκB.
La suractivation constitutive de NFκB observée chez les patients atteints de lymphome de Hodgkin, est un phénomène décrit comme intervenant dans le processus tumoral du LH. Par conséquent, dans les cas de LH associés à l'HHV-6B, cette suractivation constitutive de NFκB pourrait être accentuée par la production de protéines virales, notamment DR7 et U3, par l'HHV-6B.
CONCLUSION GENERALE ET PERSPECTIVES
1. Conclusion générale :
Nous avons pu tirer un certain nombre de conclusions de ce travail de thèse :
- Le virus HHV-6 est présent dans 35,1% des patients atteints de lymphome de Hodgkin avec une grande majorité de type B (92,3%). Les différents travaux de la littérature montrent des taux de détection très variables selon l'étude, pouvant aller de 12% à plus de 80% de détection. Ces différents résultats doivent dépendre du nombre de patients, de l'âge, des sex-ratios, et des sous-types de la pathologie. Dans notre étude, la majorité des patients infectés sont atteints du lymphome de Hodgkin de sous-type scléro-nodulaire et sont plutôt jeunes. La charge virale présente une distribution allant de 100 à 864 640 copies virales/κg d'ADN, avec une médiane autour de 500 copies /κg d'ADN, ce qui pourrait être en faveur d'un processus réplicatif dans les adénopathies de patients atteints de lymphome de Hodgkin. Il est, néanmoins, difficile d'établir le seuil du nombre de copies au-delà duquel on peut considérer que le virus se trouve dans un état réplicatif. De plus, ce seuil serait valable pour une seule technique et devrait être réévalué pour chaque technique. Par conséquent, l'état réplicatif du virus sera vérifié prochainement par la recherche des ARNm viraux dans les adénopathies de patients atteints de LH au cours d'un travail prospectif.
- Les patients atteints de lymphome de Hodgkin et porteurs de séquences d'ADN génomiques de l'HHV-6 sont, dans notre étude, rarement co-infectés par l'EBV. L'EBV n'est, par conséquent probablement pas impliqué dans les pathologies des patients de notre étude.
- Les exons 5 à 8 du gène humain de la p53, correspondant à la zone de liaison de cette protéine à l'ADN, ne portent aucune mutation, après séquençage à partir des adénopathies de la population de patients atteints de lymphome de Hodgkin. Ils semblent donc que la protéine p53 soit présente dans la cellule dans sa forme sauvage sans altération de ses fonctions due à des mutations. Les mutations du gène de la p53, signalées dans environ 50% des cancers primitifs, ne semblent pas impliquées dans la lymphomogenèse du lymphome de Hodgkin. Néanmoins, des altérations des fonctions de cette protéine peuvent exister indépendamment des mutations sur son gène.
- Le gène viral DR7 de l'HHV-6 décrit
comme oncogène, est très conservé dans les adénopathies
des patients atteints de lymphome ainsi que dans la salive des sujets sains. Il
ne présente aucune mutation associée au lymphome de Hodgkin. Par
contre, nous avons détecté quelques mutations présentes aussi
bien chez les patients que chez les sujets sains par rapport au gène DR7
de la souche Z29 en culture de l'HHV-6B, montrant que cette souche sauvage ne
semble pas très représentative des souches cliniques. La séquence
protéique de DR7 est, par conséquent, identique dans ces différentes
pathologies, impliquant une fonction conservée de cette protéine.
Nous avons également pu vérifier la faible homologie entre les gènes DR7 de l'HHV-6A et de l'HHV-6B. En effet, la majeure partie des génomes de l'HHV-6A et B semble colinéaire, mais il existe tout de même des zones moins homologues. Des différences significatives existeraient pour certains gènes régulateurs comme DR7, qui pourraient expliquer certaines différences caractéristiques entre les types viraux A et B (7, 8). - La protéine virale DR7 du type B a la capacité de se lier
à la protéine cellulaire humaine p53 permettant de penser que, si
DR7 est impliqué dans la lymphomogenèse de cette pathologie, ce
n'est pas par son gène (mutation) mais par le pouvoir oncogénique
de sa protéine. La liaison et l'inhibition de la protéine p53 constituent
une des stratégies les plus employées par les virus à ADN
impliqués dans des processus de cancérisation. Nous avons fait l'hypothèse
que si les protéines DR7A et B se liaient toutes les deux à l'ADN,
il se pouvait que ce soit sur la zone commune de ces deux protéines. Néanmoins,
elles peuvent avoir des sites de liaison différents. C'est la raison pour
laquelle de plus amples manipulations doivent être réalisées
afin de déterminer la zone exacte de liaison de la p53 sur DR7. De même,
il faut vérifier que la protéine DR7B va bien se lier comme DR7A
sur la zone de liaison à l'ADN de la p53 afin d'inhiber ses fonctions.
Si DR7B se fixe sur une zone différente, elle peut ne pas gêner la
p53 qui gardera ses fonctions intactes. Finalement, si les deux protéines
virales se fixent sur la p53 au niveau de la zone commune de leur séquence
protéique, il est possible que l'affinité de liaison soit différente
du fait de la présence de nombreuses mutations sur cette zone. De même,
il serait important de savoir dans quelle proportion la protéine DR7 est
exprimée dans la cellule infectée, de façon à estimer
si le niveau d'expression in vivo est suffisant pour entraîner
une inhibition de la p53 qui est fortement exprimée dans les cellules dont
l'ADN est endommagé.
Un grand nombre de virus à ADN intervient dans le phénomène de transformation cellulaire par l'inhibition de protéines capitales pour le cycle cellulaire comme la p53. Ces virus codent également des protéines capables de lier et d'inhiber d'autres protéines importantes pour le contrôle du cycle cellulaire comme la pRb ou la p21Waf1/Cip1. DR7 n'étant pas capable de se lier à ces protéines, il est probable que d'autres protéines virales interviennent dans ces processus. - Le virus HHV-6B a la capacité
d'augmenter la transcription des deux sous-unités p50 et p65 du complexe
NFκB et d'augmenter l'activité de ce dernier.
Les protéines virales DR7 et U3 interviennent certainement dans ce processus
de transactivation, mais nous avons montré que lors de l'infection virale,
l'augmentation des sous-unités p50 et p65 débutait à partir
de 8 heures. Or les protéines DR7 et U3 ne seraient présentes qu'à
partir de 18 heures. Il est, par conséquent, fort probable que des protéines
virales différentes interviennent en amont et que DR7 et U3 prennent le
relais.
Ce processus de transactivation de NFκB entraîne, dans les cellules, des messages importants de survie cellulaire pouvant induire une prolifération anarchique des cellules. En effet, NFκB a la capacité de transactiver la transcription de gènes anti-apoptotiques, permettant d'inhiber la cascade des caspases, protégeant ainsi la cellule de l'apoptose. Le phénomène d'activation constitutive du facteur de transcription NFκB associé au lymphome de Hodgkin pourrait être accentué dans le cas de l'infection à l'HHV-6B. Le virus, trouvé au niveau des adénopathies des patients, semble être dans un état réplicatif. Il est probable que les synthèses des protéines virales aient lieu, permettant d'avoir ainsi une grande quantité d'oncoprotéines virales. Des techniques de rétro-transcriptions et de PCR in situ, réalisées sur coupe d'adénopathies de patients atteints de LH avaient été débutées, afin de détecter la présence des ARNm de DR7 et U3 dans la cellule de Reed-Sternberg. Mais les différentes étapes de congélations des biopsies ont rendu la détection des ARNm impossible du fait de leurs altérations. Cette recherche sera également réinitialisée lors de l'étude d'adénopathies de patients atteints de LH envisagée en prospectif.
En conclusion, nous avons mis en évidence une probable implication du virus HHV-6B dans la lymphomogenèse du lymphome de Hodgkin et ce, par au moins deux mécanismes différents :
- la séquestration de la protéine suppresseur de tumeur p53,
- la suractivation de l'activité du complexe cellulaire NFκB (Figure 21).
Mais il faut rester prudent sur cette implication. D'autres herpèsvirus ont également la capacité de transactiver NFκB comme le CMV ou l'EBV. Dans le cas du CMV, malgré cette dérégulation, et le fait que des protéines virales aient également la capacité d'inhiber la p53 et la pRb, ce virus n'a été associé à aucun cancer humain. Il présenterait seulement un potentiel oncogène in vitro, capable de transformer des lignées cellulaires.
D'autre part, il est évident que l'HHV-6 ne déclenche pas l'apparition du lymphome car dans plus de 60% des cas de LH, il n'est pas détecté. Par conséquent, il faudrait considérer ce virus comme opportuniste, profitant d'une situation de faiblesse du système immunitaire pour déréguler des voies de contrôle du cycle cellulaire. Dans cette hypothèse, le virus pourrait accentuer le phénomène de transformation cellulaire. Mais, l'infection par l'HHV-6 ne semble pas altérer le pronostic de survie des patients. Les traitements utilisés semblent suffisamment efficaces (taux de guérison de l'ordre de 80%) pour masquer un éventuel effet néfaste de l'HHV-6.
Au cours de ce travail, les résultats obtenus n'aboutissent pas à établir des conclusions définitives en ce qui concerne l'implication de l'HHV-6 dans le développement du lymphome de Hodgkin. Ils constituent, en revanche, un point de départ dans l'étude des mécanismes oncogènes de l'HHV-6 dans le lymphome de Hodgkin.
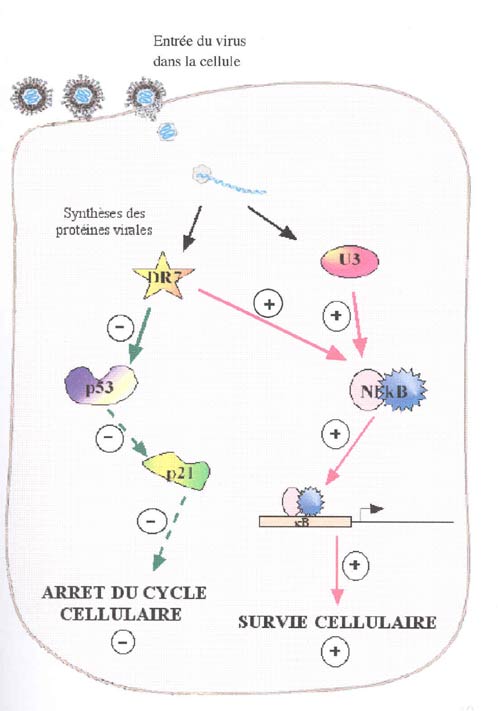
La protéine virale DR7 séquestre la p53 et par conséquent pourrait empêcher l'arrêt du cycle cellulaire. Les protéines U3 et DR7, sur-activant NFκB, pourraient entraîner une augmentation des signaux de survie cellulaire ; ces deux processus pouvant aboutir à une prolifération anarchique des cellules.
2. Perspectives :
-
Comme nous l'avons précisé au cours de ce travail, une étude
prospective sur des adénopathies de patients atteints de lymphome de Hodgkin
est envisagée afin de rechercher, entre autre, des ARNm viraux (précoces
et tardifs) pour vérifier l'état réplicatif du virus dans
les adénopathies de ces patients.
Nous avons montré que l'action oncogénique de l'HHV-6B pouvait notamment provenir de l'activité des protéines virales DR7 et U3. De ce fait, plusieurs études peuvent être envisagées pour poursuivre le travail dans cette optique : - La synthèse de ces protéines virales DR7 et U3, afin de déterminer leur fonction au sein du cycle viral. Le système Rapid Translation System 500 (RTS 500, Roche), ayant été essayé sans succès, un système de synthèse de protéines, par baculovirus en cellules d'insectes est en cours de développement dans le laboratoire.
- La détermination des mécanismes employés par ces protéines pour agir sur la transactivation des gènes cellulaires. Des expériences de gel retard (EMSA Electrophoresis mobility shift assay) sont envisagées pour élucider ces questions.
- La recherche d'autres cibles cellulaires à ces protéines virales avec des essais de coimmunoprécipitation et de double hybride.
- La recherche du virus HHV-6B et la détection de l'expression de ces deux protéines dans les cellules de Reed-Sternberg. Pour cela, des micromanipulations doivent être réalisées sur des infiltrats cellulaires provenant de biopsies de patients atteints de lymphome de Hodgkin. Mais il s'agit de techniques que nous ne pratiquons pas au laboratoire. Nous envisageons, par conséquent, de collaborer avec une équipe de recherche maîtrisant ce genre de technologies.
- Si le virus HHV-6 est trouvé dans les cellules de RS, il serait intéressant de vérifier si ce virus est capable d'induire les phosphorylations constitutives des facteurs de transcription STAT3 et STAT6 décrites comme associées au phénomène de cancérisation du lymphome de Hodgkin. De même, l'HHV-6 ne pourrait-il pas favoriser le processus de méthylation de la zone promotrice du gène de l'inhibiteur des kinases p16 trouvée dans les cellules de RS ?
- Le génome de l'HHV-6 codant certainement plusieurs protéines virales oncogènes, la recherche de celles capables d'inhiber les fonctions des protéines pRb et p21Waf1/Cip1 serait également intéressante.
Finalement, un travail important reste à fournir afin de déterminer exactement l'implication de ce virus dans le développement du lymphome de Hodgkin.
VALORISATION DES COMPETENCES
1. Présentation du travail de thèse :
Pendant ce travail de thèse, nous avons étudié l'implication de l'herpèsvirus humain de type 6 (HHV-6) dans le processus de cancérisation du lymphome de Hodgkin (LH). Le LH est un cancer du système lymphatique apparaissant surtout chez des patients jeunes. L'HHV-6 est anormalement présent dans les tumeurs de certains patients atteints de LH. Nous voulions savoir si le virus pouvait intervenir dans le développement du lymphome. Nous avons travaillé sur des échantillons de ganglions lymphatiques provenant d'une population de patients atteints de LH, au sein de laquelle nous avons détecté le virus dans environ 35% des cas. Nous avons déterminé que le virus avait la capacité de déréguler la multiplication des cellules dans lesquelles il se trouvait. Dans un état physiologique, la cellule se multiplie pendant un certain temps et sous certaines conditions, ou se maintient dans un état de quiescence, puis meurt, le tout hautement contrôlé par des mécanismes cellulaires. Dans les phénomènes de cancérisation, la cellule se multiplie sans aucun contrôle aboutissant à la formation de tumeurs. Au cours de cette étude, nous avons montré que le virus HHV-6 pouvait (1) inhiber une voie de contrôle visant à arrêter la multiplication cellulaire et (2) activer une voie de signalisation permettant de stimuler la multiplication cellulaire.
Nous avons conclu de cette étude que l'infection par le virus HHV-6 pouvait probablement activer le processus de tumorisation.
2. Travail de thèse au sein de la thématique du laboratoire :
Le laboratoire qui m'a accueillie pour mon travail de thèse est principalement un laboratoire d'analyses cliniques composée d'une équipe importante de techniciens ainsi qu'un petit groupe de chercheurs.
La thématique du laboratoire de recherche est surtout basée sur l'étude des traitements antiviraux et sur l'étude des conséquences de ces traitements sur les patients et sur le virus. Notre sujet de recherche est un peu parallèle à cette thématique. Un des buts de ce travail était d'essayer de déterminer s'il est intéressant de traiter ou pas les patients atteints de LH et infectés par l'HHV-6. Si l'HHV-6 entraîne une accélération du processus de cancérisation, il serait peut-être intéressant de traiter les patients infectés par l'HHV-6 afin de freiner le développement du cancer lié au virus. L'équipe de notre laboratoire travaille sur les doses d'antiviraux entraînant une résistance du virus. La résistance, qu'elle soit bactérienne face aux antibiotiques, ou virale face aux antiviraux, est un problème capital d'actualité. Il est important de ne pas traiter des patients quand ça n'est pas nécessaire, permettant ainsi de limiter le développement de résistances chez ces micro-organismes contre les traitements utilisés.
3. Concurrence :
Le laboratoire dans lequel j'ai effectué ma thèse travaille depuis longtemps sur le virus HHV-6. Quelques équipes de recherche seulement étudient ce virus. Il existe nationalement une équipe à Paris. Par contre, il existe quelques grandes équipes à l'étranger, en Italie, en GB, au Canada, aux USA et au Japon. Mais très peu d'équipes étudient l'association cancer-virus. Il est par conséquent difficile de travailler en collaboration avec d'autres équipes. L'implication de ce virus dans le développement de certains cancers est encore très incertaine. L'HHV-6 ne présente pas un caractère oncogène très sévère, son action serait plus insidieuse, c'est la raison pour laquelle les équipes travaillant sur l'association herpèsvirus-cancer s'intéressent à des virus plus virulents comme le virus d'Epstein-Barr (EBV). Les équipes étudiant l'HHV-6 sont plus intéressées par les conséquences de l'infection chez les patients immunodéprimés, car cela représente un secteur économiquement plus fructueux pouvant aboutir à l'établissement de nouvelles thérapies. Le choix de cette thématique provient de la taille du laboratoire. En effet, l'équipe dans laquelle j'ai travaillé, est une petite équipe qui peut difficilement rivaliser avec de gros laboratoires.
Par contre, nous avons travaillé avec un certain nombre de laboratoires étrangers qui nous ont contactés afin d'utiliser dans leur propre laboratoire, des systèmes de détection que nous avions développés. Nous avons travaillé avec un laboratoire parisien, tchèque et américain, qui ont mis en pratique dans leur laboratoire un de nos systèmes de détection du virus.
4. Moyens à ma disposition :
J'ai intégré une équipe de recherche compétente dans des domaines très variés comme la biologie cellulaire et moléculaire. J'ai bénéficié des connaissances d'un grand nombre de techniciens de laboratoire ainsi que d'ingénieurs de recherche, qui m'ont formée sur différentes techniques pendant toute la durée de ma thèse. Enfin, pour les techniques que nous voulions nouvellement développer, j'ai eu la chance d'assister à des congrès, séminaires et stages ainsi que de travailler avec plusieurs autres équipes de recherche.
Secondairement, j'ai eu le privilège de travailler avec une équipe ouverte sur le plan des nouveautés me permettant de développer des systèmes très innovants, sans avoir, dans la mesure du raisonnable, de limites tant sur le plan scientifique que financier.
5. Evaluation du coût de la thèse :
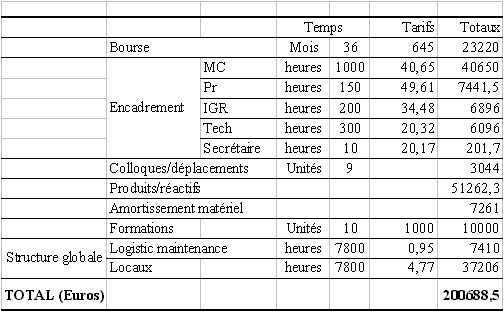
MC : Maître de conférence, Pr : Professeur, IGR : Ingénieur de Recherche, Tech : Technicien.
6. Conduite du projet :
Tout au long de ce travail de thèse, l'organisation de mon temps de travail m'incombait entièrement. Nous avions une réunion de laboratoire chaque fin de semaine pendant laquelle, tous les doctorants et chercheurs présentaient l'avancée de leurs travaux. Nous avions, par conséquent, l'avantage de gérer nous-même notre temps de travail. Par contre, nous devions partager l'équipement du laboratoire avec des techniciens dont le travail était prioritaire du fait de leur activité d'analyses médicales. Une planification d'utilisation du matériel commun a été établie à la suite de plusieurs concertations entre les différents partenaires, nous permettant une gestion du temps de travail optimal pour l'équipe de recherche, malgré la priorité de l'équipe de techniciens. Nous avons finalement réussi à établir une bonne collaboration entre les équipes de techniciens et de chercheurs.
J'ai élaboré mon travail de thèse suivant le schéma global suivant : (A) Suite à une étude bibliographique du sujet, un certain nombre de questions sont énoncées. (B) Quels sont les moyens techniques mis en oeuvre pour y répondre ? (Choix des techniques, établissement de protocoles, planification du temps de travail...) (C) Quels sont les résultats obtenus ? (D) Interprétations de ces résultats. (E) Soumissions des résultats à l'équipe de recherche lors des réunions. A partir de cette étape, plusieurs solutions sont envisageables : soit l'interprétation est correcte et l'étude est poursuivie, soit elle ne l'est pas et il faut refaire l'interprétation, ou réaliser de nouvelles manipulations techniques. (F) Etablissement des conclusions. (G) Ecriture de l'article.
Au cours des différentes études menées pendant ce travail, j'ai établi des collaborations étroites avec les services techniques des entreprises qui commercialisaient les systèmes ou produits utilisés. En général, lors de ces partenariats, chaque participant trouve un avantage. En effet, nous travaillons avec les personnes ayant développés les systèmes que nous utilisons, nous permettant de gagner du temps, mais ces personnes cherchaient également à connaître les évolutions ou les comportements de leurs systèmes dans telle ou telle condition.
J'ai présenté, au cours de ma thèse, mon travail de recherche dans des congrès nationaux et internationaux. En effet, nous avons exposé un grand nombre de travaux préliminaires ou finaux avant publication, nous permettant, dans un premier temps, de nous imposer des dates limites dans notre travail, mais aussi de nous faire connaître, et par la même occasion, de faire critiquer notre travail. Il est parfois difficile de conserver un regard objectif sur un travail dans lequel nous sommes très impliqués. Et finalement, nous soumettions les travaux dans différentes revues. Les articles soumis ont rarement été acceptés directement, ils ont souvent été acceptés sous réserve de modifications mais également refusés. Il faut alors tenir compte des critiques émises et soumettre à nouveau l'article corrigé dans d'autres revues.
Une fois les articles relatifs à la thèse soumis ou acceptés, j'ai envisagé de soutenir ma thèse à la fin de mes trois années. Il m'a paru plus intéressant de ne pas prolonger ce travail d'une quatrième année, de façon à pouvoir envisager plus rapidement mon avenir. Il est très difficile de mener en parallèle un travail de thèse très prenant avec son insertion professionnelle. Nous avons la chance, à Limoges, d'appartenir à une école doctorale très dynamique, nous permettant de réaliser un grand nombre de stages dans des domaines très variés, et notamment sur l'avenir professionnel des docteurs. Nous avons eu l'opportunité de bénéficier de conseils de personnels des points relais cadre de l'ANPE ainsi que des personnes travaillant dans des cabinets de recrutement. J'ai, par conséquent, pu élaborer mon projet de recherche d'emploi à l'aide de tous les conseils obtenus lors de ces stages, mais aussi lors de mes diverses rencontres avec des professionnels du secteur.
7. Compétences autres que scientifiques :
Au cours de ce travail de thèse, j'ai développé un certain nombre de compétences autres que scientifiques. La gestion de projet est certainement la principale. En effet, je n'avais jamais réalisé un projet aussi important. Il a fallu résoudre un problème dans un délai donné et avec des impératifs de moyens techniques, de personnels et financiers. Ceci impliquait une gestion serrée du timing des manipulations techniques sur du court, et du long terme. Il fallait envisager le temps de travail maximal à fournir pour obtenir tel ou tel renseignement, mais aussi le temps que l'obtention de l'ensemble des résultats prendrait jusqu'à l'écriture et la soumission de l'article qui en découlait. Il ne fallait surtout pas perdre de vue le délai final de trois ans.
Au cours de ce travail, j'ai eu le privilège d'encadrer des étudiants de différents niveaux scientifiques. Il a fallu organiser leur temps de travail en fonction du mien, leur permettant de devenir de plus en plus autonomes. Il faut à ce moment-là établir une véritable gestion de personnel et réaliser des formations aussi bien théoriques que pratiques.
L'ensemble des dépenses effectuées pour mon travail devait être validé par mon directeur de thèse. L'équipe de chercheurs disposait d'un budget annuel. Nous devions, par conséquent, établir dès la fin de l'année une planification quasiment exhaustive de l'ensemble des techniques que nous envisagions pour l'année suivante afin de partager le plus équitablement cette somme en fonction des besoins de chacun.
Durant ce travail de thèse au sein de cette équipe composée majoritairement de techniciens, j'ai appris la diplomatie nécessaire à toute cohabitation afin de permettre l'établissement d'un climat de travail efficace, chaleureux et amical.
J'ai également développé mes capacités à convaincre. Il a fallu que je défende mon projet de recherche, mais aussi que les changements d'axes que j'envisageais, ainsi que les techniques employées pour résoudre les problèmes. Chaque proposition faisait l'objet d'une étude par les différents directeurs qui était, soit acceptée, soit refusée. En cas de refus, des discussions s'ouvraient nous permettant de défendre notre projet.
J'ai également pu améliorer ma pratique de l'anglais grâce à de nombreux stages d'anglais organisés par l'Ecole Doctorale, mais aussi par des séjours aux USA lors de congrès internationaux. J'ai également eu le privilège de présenter oralement en anglais mes travaux de recherche lors de ces congrès.
J'ai aussi appris à utiliser des logiciels appliqués à la biologie moléculaire tels que DNA Strider 1.2, Geneworks 2.5, Sequence Navigator, Sequence analysis 3.4, Mac Vector 7.0. J'ai également appris à maîtriser l'utilisation des banques de données de biologie moléculaire sur Internet comme GENBANK mais aussi BLAST, les banques de données bibliographiques (PubMed) ainsi que des moteurs de recherche moins spécialisés (Google, Altavista, Metacrawler...). J'ai pu améliorer ma pratique des logiciels de traitements de texte (Word, Powerpoint), de tableur (Excel), des logiciels de dessins (ClarisDraw) et de retouche de photos (PhotoShop 5.0) mais aussi des logiciels de traitements de données bibliographiques (EndNote).
8. Conclusions :
Mon travail de thèse pourrait être divisé en plusieurs phases : au départ, le sujet de l'étude était pré-établi ainsi que les questions posées et les moyens pour y répondre ; mon rôle était seulement de réaliser ce qui était prévu. Dans un deuxième temps, mon tuteur continuait à faire évoluer mon sujet de recherche, mais je devais trouver les moyens à mettre en place pour répondre aux différentes questions posées. Et dans un troisième temps, suivant les résultats obtenus lors des phases précédentes, j'ai recadré moi-même mon sujet et développé les systèmes nécessaires. Globalement, j'ai l'impression d'avoir commencé mon travail de thèse comme technicienne de laboratoire et de l'avoir achevé comme chercheur à part entière.
Les résultats obtenus lors de ce travail m'ont permis de publier des articles scientifiques de revues ainsi que des articles de recherche plus fondamentaux. J'ai acquis des connaissances dans le domaine de la biologie moléculaire, cellulaire et dans la virologie. J'ai appris un grand nombre de techniques et j'ai pu en développer d'autres dont certaines ont été adoptées pour un usage clinique, permettant un échange bénéfique avec le laboratoire. Au cours de mon travail, mon tuteur m'a permis de me spécialiser dans un domaine particulier, et d'acquérir des connaissances sur un système innovant (PCR en temps réel). Le laboratoire a, par la suite, acquis cette technologie, ce qui m'a permis de développer tous les systèmes nécessaires à mon travail de thèse. En contre-partie, les systèmes développés, ont été adaptés pour la recherche clinique et j'ai eu l'occasion de former une partie du personnel de façon à les rendre complètement autonomes.
J'ai également travaillé avec des équipes de recherche très différentes, qui m'ont permis de développer dans notre laboratoire des techniques que nous ne maîtrisions pas. J'ai réalisé, au cours de ces collaborations, que le dialogue et surtout la sincérité étaient des notions capitales pour un travail sain et efficace entre différentes équipes. Je crois avoir établi des contacts solides tant sur le plan professionnel que personnel. J'ai éprouvé, un grand plaisir à travailler avec des gens très différents.
J'ai également eu le privilège d'assister à un grand nombre de congrès et séminaires qui m'ont permis d'établir de sérieux contacts avec des scientifiques mais aussi avec des professionnels des biotechnologies. J'ai bénéficié de ces contacts à différentes d'occasions. De même, j'ai présenté mon travail à plusieurs reprises lors de ces congrès et j'ai appris, d'une part, à être jugée, et d'autre part à partager mes connaissances. J'ai tout particulièrement apprécié les congrès internationaux permettant de rencontrer des personnes très différentes et de voir, de façon surprenante, que la culture des personnes influençait énormément leur mode de réflexion et leur façon d'aborder les problèmes scientifiques.
Nous avons également établi des collaborations efficaces avec certains laboratoires suite à ces différents congrès scientifiques. J'ai eu la chance d'être aidée par des équipes qui ont pu me fournir des réactifs que je ne pouvais pas me procurer. Inversement, nous avons également fourni certains systèmes de détection développés dans le laboratoire à d'autres laboratoires intéressés par l'obtention d'une méthode clef en main. En dehors de mon sujet de thèse, nous avons également établi une collaboration avec une équipe de virologues parisiens. Ils possédaient une série d'échantillons provenant de patients atteints d'une pathologie dans laquelle ils supposaient une implication de l'HHV-6, mais ne possédaient pas les outils nécessaires à cette démonstration. Ils nous ont contactés, suite à un congrès, et nous avons établi ensemble une stratégie de recherche. Nous avons appliqué nos systèmes de détection sur leurs échantillons. Cela leur a permis de publier deux articles différents dans des revues cliniques bien classées. Nous avons été associées, mon tuteur et moi, aux deux articles, pour un temps passé relativement court. Dans ce cas-là, l'association a été très productive, mais ce n'est pas toujours le cas. Quelquefois, les hypothèses avancées ne sont pas vérifiées par la pratique et le projet échoue. Il faut par conséquent faire un choix au départ afin de ne pas perdre trop de temps en collaborations improductives. De même, pendant la thèse, il ne faut pas perdre de vue l'essentiel qui est de mener à bien un sujet de recherche. Or tout au long de ce temps, nous sommes sollicités par nos tuteurs, par nos encadrants, par des collaborations éventuelles, ou par nous-même, pour travailler sur des sujets très intéressants mais différents de la thématique de la thèse. Il faut, par conséquent, essayer de conserver le cap du travail principal, tout en réalisant de façon occasionnelle des travaux ne s'intégrant pas directement dans le travail de thèse.
9. Projet professionnel :
La thèse est à mon avis une excellente expérience personnelle. Elle permet de se rendre compte de ses propres limites et de se responsabiliser. En effet, d'un point de vue scientifique, nous devons publier des articles relatant des faits expérimentaux et non pas des interprétations erronées de mauvaises techniques ou des techniques employées dans un mauvais contexte. D'autre part, nous avons à notre disposition des sommes importantes pour réaliser notre étude. Il faut apprendre à les gérer du mieux possible afin de ne pas gaspiller cet argent. Et finalement, notre formation de docteur coûte très cher à la société. Ces différents paramètres obligent l'étudiant à se responsabiliser et à prendre conscience de l'importance du sérieux qu'il doit amener à son travail.
Secondairement, je considère la thèse comme une véritable expérience professionnelle. En effet, l'étudiant doit, tout au long de la thèse, gérer des projets, des budgets et du personnel tout comme un cadre doit le faire dans son entreprise. Le laboratoire dans lequel j'ai travaillé est composé d'une quarantaine de techniciens, d'une dizaine de biologistes et d'une douzaine de chercheurs. Il pourrait être, par conséquent, comparé à une petite entreprise avec toutes ses exigences d'organisation du travail, de budget et de personnel.
J'envisage d'intégrer le monde de l'entreprise pour différentes raisons. Premièrement, les possibilités de carrière dans la recherche académique sont de plus en plus réduites. Et surtout, je me suis aussi rendu compte au cours de ce travail que j'appréciais de faire des choses très différentes. La recherche m'a passionnée et j'ai énormément apprécié de pouvoir présenter les données dans des congrès me permettant d'entrer en contact avec des personnes très différentes. Il me semble que j'apprécierais tout particulièrement un poste me permettant de mettre en pratique mes compétences scientifiques mais présentant également un aspect commercial.